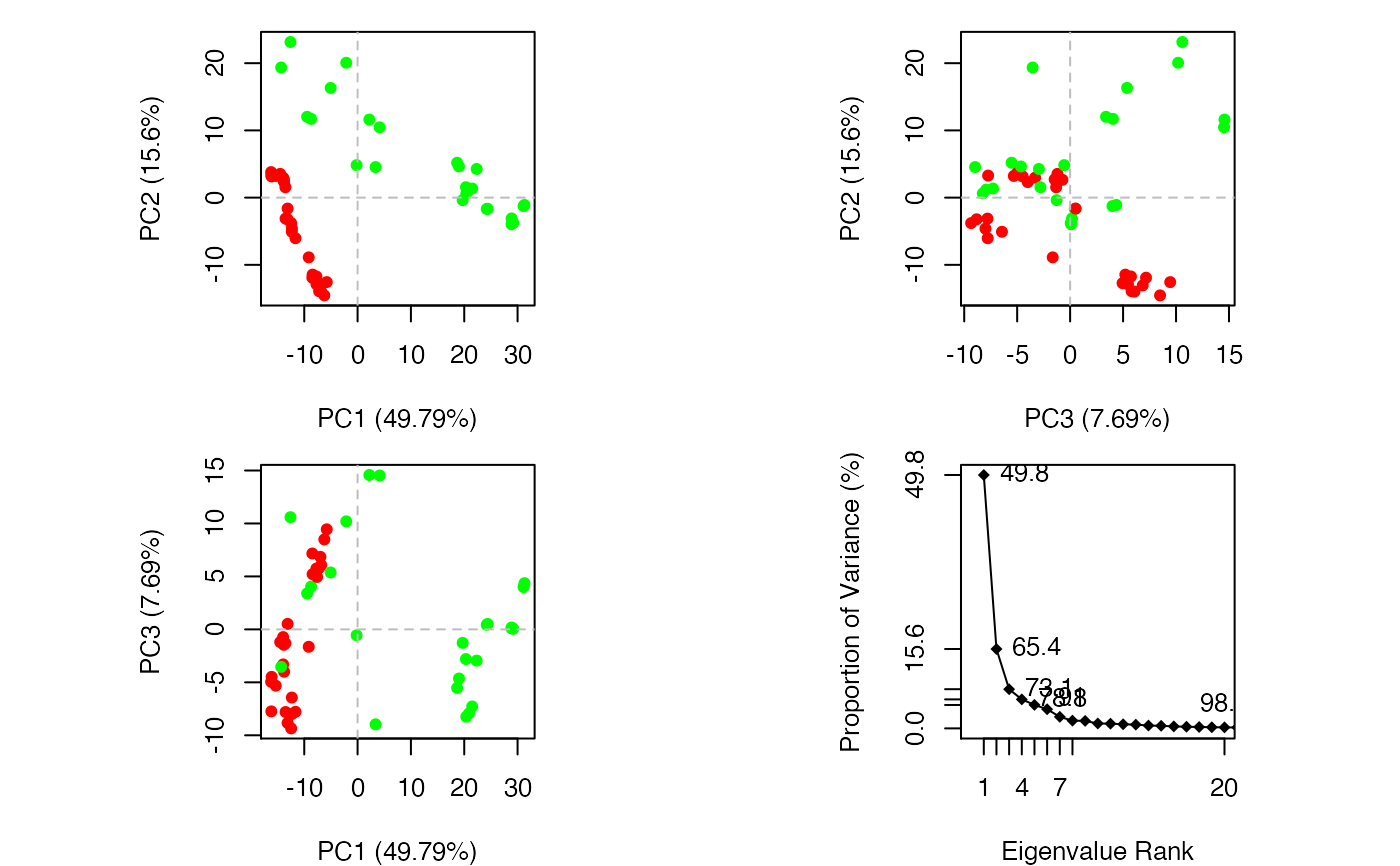
Principal Component Analysis
pca.pdbs.RdPerforms principal components analysis (PCA) on an ensemble of PDB structures.
# S3 method for pdbs pca(pdbs, core.find = FALSE, fit = FALSE, ...)
Arguments
| pdbs | an object of class |
|---|---|
| core.find | logical, if TRUE core.find() function will be called to find core positions and coordinates of PDB structures will be fitted based on cores. |
| fit | logical, if TRUE coordinates of PDB structures will be fitted based on all CA atoms. |
| ... | additional arguments passed to the method |
Details
The function pca.pdbs is a wrapper for the function
pca.xyz, wherein more details of the PCA procedure
are documented.
Value
Returns a list with the following components:
eigenvalues.
eigenvectors (i.e. the variable loadings).
scores of the supplied data on the pcs.
the standard deviations of the pcs.
the means that were subtracted.
References
Grant, B.J. et al. (2006) Bioinformatics 22, 2695--2696.
Author
Barry Grant, Lars Skjaerven and Xin-Qiu Yao
See also
Examples
attach(transducin) #-- Do PCA ignoring gap containing positions pc.xray <- pca(pdbs) # Plot results (conformer plots & scree plot) plot(pc.xray, col=annotation[, "color"])detach(transducin)