PDB structure manipulation and analysis
Lars Skjaerven, Xin-Qiu Yao & Barry J. Grant
Bio3D_pdb.RmdIntroduction
Bio3D1 is a family of related R packages containing utilities for the analysis of biomolecular structure, sequence and trajectory data (Grant et al. 2006; Skjaerven et al. 2014). Features include the ability to read and write biomolecular structure, sequence and dynamic trajectory data, perform atom selection, re-orientation, superposition, rigid core identification, clustering, distance matrix analysis, conservation analysis, normal mode analysis, principal component analysis, and protein structure and correlation network analysis. Bio3D takes advantage of the extensive graphical and statistical capabilities of the R environment and thus represents a useful framework for exploratory analysis of structural data.
New to R?
There are numerous on–line resources that can help you get started using R effectively. A number of these can be found from the main R website at http://www.r-project.org. We particularly like the following:
-
R primers from RStudio: an interactive R tutorial in your web browser,
-
DataCamp’s Introduction to R: Learn by doing in your web browser (requires free registration),
-
Introduction to R and RStudio video series: Some of Barry’s course material from UC San Diego,
- Offical introduction to R: The offical R manual.
Using this vignette
The aim of this document, termed a vignette2 in R parlance, is to provide a brief introduction to PDB structure manipulation and analysis with the Bio3D core package (bio3d). A number of other Bio3D package vignettes and tutorials are available online at http://thegrantlab.org/bio3d. In particular, detailed instructions for obtaining and installing the Bio3D packages on various platforms can be found in the Installing Bio3D Vignette. Note that this vignette was generated using bio3d version 2.4.1.9000.
Getting started
Start R (type R at the command prompt or, on Windows, double click on the R icon) and load the bio3d package by typing library(bio3d) at the R console prompt.
Then use the command lbio3d() or help(package=bio3d) to list the functions within the package and help(FunctionName) to obtain more information about an individual function.
# List of bio3d functions with brief description help(package=bio3d) # Detailed help on a particular function, e.g. 'pca.xyz' help(pca.xyz)
To search the help system for documentation matching a particular word or topic use the command help.search("topic"). For example, help.search("pdb")
help.search("pdb")
Typing help.start() will start a local HTML interface to the R documentation and help system. After initiating help.start() in a session the help() commands will open as HTML pages in your web browser.
bio3d functions and their typical usage
The bio3d package consists of input/output functions, conversion and manipulation functions, analysis functions, and graphics functions all of which are fully documented both online and within the R help system introduced above.
To better understand how a particular function operates it is often helpful to view and execute an example. Every function within the Bio3D packages is documented with example code that you can view by issuing the help() command.
Running the command example(function) will directly execute the example for a given function. In addition, a number of longer worked examples are available as Tutorials on the Bio3D website.
example(plot.bio3d)
Working with individual PDB files
Protein Data Bank files (or PDB files) are the most common format for the distribution and storage of high-resolution biomolecular coordinate data. The bio3d package contains functions for the reading (read.pdb(), read.fasta.pdb(), get.pdb(), convert.pdb(), basename.pdb()), writing (write.pdb()) and manipulation (trim.pdb(), cat.pdb(), pdbsplit(), atom.select(), pdbseq()) of PDB files. Indeed numerous Bio3D analysis functions are intended to operate on PDB file derived data (e.g. blast.pdb(), chain.pdb(), nma.pdb(), pdb.annotate(), pdbaln(), pdbfit(), struct.aln(), dssp(), pca.pdbs() etc.)
At their most basic, PDB coordinate files contain a list of all the atoms of one or more molecular structures. Each atom position is defined by its x, y, z coordinates in a conventional orthogonal coordinate system. Additional data, including listings of observed secondary structure elements, are also commonly (but not always) detailed in PDB files.
Read a PDB file
To read a single PDB file with Bio3D we can use the read.pdb() function. The minimal input required for this function is a specification of the file to be read. This can be either the file name of a local file on disc or the RCSB PDB identifier of a file to read directly from the on-line PDB repository. For example to read and inspect the on-line file with PDB ID 4q21:
pdb <- read.pdb("4q21")
## Note: Accessing on-line PDB fileTo get a quick summary of the contents of the pdb object you just created you can issue the command print(pdb) or simply type pdb (which is equivalent in this case):
pdb
##
## Call: read.pdb(file = "4q21")
##
## Total Models#: 1
## Total Atoms#: 1447, XYZs#: 4341 Chains#: 1 (values: A)
##
## Protein Atoms#: 1340 (residues/Calpha atoms#: 168)
## Nucleic acid Atoms#: 0 (residues/phosphate atoms#: 0)
##
## Non-protein/nucleic Atoms#: 107 (residues: 80)
## Non-protein/nucleic resid values: [ GDP (1), HOH (78), MG (1) ]
##
## Protein sequence:
## MTEYKLVVVGAGGVGKSALTIQLIQNHFVDEYDPTIEDSYRKQVVIDGETCLLDILDTAG
## QEEYSAMRDQYMRTGEGFLCVFAINNTKSFEDIHQYREQIKRVKDSDDVPMVLVGNKCDL
## AARTVESRQAQDLARSYGIPYIETSAKTRQGVEDAFYTLVREIRQHKL
##
## + attr: atom, xyz, seqres, helix, sheet,
## calpha, remark, callNote that the attributes (+ attr:) of this object are listed on the last couple of lines. To find the attributes of any such object you can use:
attributes(pdb)
## $names
## [1] "atom" "xyz" "seqres" "helix" "sheet" "calpha" "remark" "call"
##
## $class
## [1] "pdb" "sse"To access these individual attributes we use the dollar-attribute name convention that is common with R list objects. For example, to access the atom attribute or component use pdb$atom:
head(pdb$atom)
## type eleno elety alt resid chain resno insert x y z o b
## 1 ATOM 1 N <NA> MET A 1 <NA> 64.080 50.529 32.509 1 28.66
## 2 ATOM 2 CA <NA> MET A 1 <NA> 64.044 51.615 33.423 1 29.19
## 3 ATOM 3 C <NA> MET A 1 <NA> 63.722 52.849 32.671 1 30.27
## 4 ATOM 4 O <NA> MET A 1 <NA> 64.359 53.119 31.662 1 34.93
## 5 ATOM 5 CB <NA> MET A 1 <NA> 65.373 51.805 34.158 1 28.49
## 6 ATOM 6 CG <NA> MET A 1 <NA> 65.122 52.780 35.269 1 32.18
## segid elesy charge
## 1 <NA> N <NA>
## 2 <NA> C <NA>
## 3 <NA> C <NA>
## 4 <NA> O <NA>
## 5 <NA> C <NA>
## 6 <NA> C <NA># Print $atom data for the first two atoms pdb$atom[1:2, ]
## type eleno elety alt resid chain resno insert x y z o b
## 1 ATOM 1 N <NA> MET A 1 <NA> 64.080 50.529 32.509 1 28.66
## 2 ATOM 2 CA <NA> MET A 1 <NA> 64.044 51.615 33.423 1 29.19
## segid elesy charge
## 1 <NA> N <NA>
## 2 <NA> C <NA># Print a subset of $atom data for the first two atoms pdb$atom[1:2, c("eleno", "elety", "x","y","z")]
## eleno elety x y z
## 1 1 N 64.080 50.529 32.509
## 2 2 CA 64.044 51.615 33.423# Note that individual $atom records can also be accessed like this pdb$atom$elety[1:2]
## [1] "N" "CA"# Which allows us to do the following (see Figure 1.) plot.bio3d(pdb$atom$b[pdb$calpha], sse=pdb, typ="l", ylab="B-factor")

Residue temperature factors for PDB ID 4q21 with secondary structure element (SSE) annotation in marginal regions plotted with function plot.bio3d()
Note that the main xyz coordinate attribute is a numeric matrix with 3N columns (each atom has three values x, y and z). The number of rows here correspond to the number of models in the PDB file (typically one for X-ray structures and multiple for NMR structures).
# Print a summary of the coordinate data in $xyz pdb$xyz
##
## Total Frames#: 1
## Total XYZs#: 4341, (Atoms#: 1447)
##
## [1] 64.08 50.529 32.509 <...> 74.159 76.923 41.999 [4341]
##
## + attr: Matrix DIM = 1 x 4341# Examine the row and column dimensions dim(pdb$xyz)
## [1] 1 4341pdb$xyz[ 1, atom2xyz(1:2) ]
## [1] 64.080 50.529 32.509 64.044 51.615 33.423Side-note: The ‘pdb’ class
Objects created by the read.pdb() function are of class “pdb”. This is recognized by other so called generic Bio3D functions (for example atom.select(), nma(), print(), summary() etc.). A generic function is a function that examines the class of its first argument, and then decides what type of operation to perform (more specifically it decides which specific method to dispatch to). So for example, the generic atom.select() function knows that the input is of class “pdb”, rather than for example an AMBER parameter and topology file, and will act accordingly.
A careful reader will also of noted that our “pdb” object created above also has a second class, namely “sse” (see the output of attributes(pdb) or class(pdb)). This stands for secondary structure elements and is recognized by the plot.bio3d() function to annotate the positions of major secondary structure elements in the marginal regions of these plots (see Figure 1). This is all part of the R S3 object orientation system. This S3 system os used throughout Bio3D to simplify and facilitate our work with these types of objects.
Atom selection
The bio3d atom.select() function is arguably one of the most challenging for newcomers to master. It is however central to PDB structure manipulation and analysis. At its most basic, this function operates on PDB structure objects (as created by read.pdb()) and returns the numeric indices of a selected atom subset. These indices can then be used to access the $atom and $xyz attributes of PDB structure related objects.
For example to select the indices for all C-alpha atoms we can use the following command:
# Select all C-alpha atoms (return their indices) ca.inds <- atom.select(pdb, "calpha") ca.inds
##
## Call: atom.select.pdb(pdb = pdb, string = "calpha")
##
## Atom Indices#: 168 ($atom)
## XYZ Indices#: 504 ($xyz)
##
## + attr: atom, xyz, callNote that the attributes of the returned ca.inds from atom.select() include both atom and xyz components. These are numeric vectors that can be used as indices to access the corresponding atom and xyz components of the input PDB structure object. For example:
# Print details of the first few selected atoms head( pdb$atom[ca.inds$atom, ] )
## type eleno elety alt resid chain resno insert x y z o b
## 2 ATOM 2 CA <NA> MET A 1 <NA> 64.044 51.615 33.423 1 29.19
## 10 ATOM 10 CA <NA> THR A 2 <NA> 62.439 54.794 32.359 1 28.10
## 17 ATOM 17 CA <NA> GLU A 3 <NA> 63.968 58.232 32.801 1 30.95
## 26 ATOM 26 CA <NA> TYR A 4 <NA> 61.817 61.333 33.161 1 23.42
## 38 ATOM 38 CA <NA> LYS A 5 <NA> 63.343 64.814 33.163 1 21.34
## 47 ATOM 47 CA <NA> LEU A 6 <NA> 61.321 67.068 35.557 1 18.99
## segid elesy charge
## 2 <NA> C <NA>
## 10 <NA> C <NA>
## 17 <NA> C <NA>
## 26 <NA> C <NA>
## 38 <NA> C <NA>
## 47 <NA> C <NA># And selected xyz coordinates head( pdb$xyz[, ca.inds$xyz] )
## [1] 64.044 51.615 33.423 62.439 54.794 32.359In addition to the common selection strings (such as ‘calpha’ ‘cbeta’ ‘backbone’ ‘protein’ ‘notprotein’ ‘ligand’ ‘water’ ‘notwater’ ‘h’ and ‘noh’) various individual atom properties can be used for selection.
# Select chain A a.inds <- atom.select(pdb, chain="A") # Select C-alphas of chain A ca.inds <- atom.select(pdb, "calpha", chain="A") # We can combine multiple selection criteria to return their intersection cab.inds <- atom.select(pdb, elety=c("CA","CB"), chain="A", resno=10:20)
Inverse selection
The atom.select() function also contain options to inverse the final selection. For example, including the argument inverse=TRUE in the statement below will select all atoms except the water atoms:
# Select all atoms except waters nowat.inds <- atom.select(pdb, "water", inverse=TRUE)
Combining selections
The operator argument of atom.select() determines how to combine the individual selection statements. The operator is by default "AND", which specifies that the final selection is combined by intersection of the individual selection statements. In contrast, by setting operator="OR" the final selection is determined by the union of the individual statements. For example, the following selection will select all protein atoms as well as those of any residue named GDP:
# Select protein + GDP sele <- atom.select(pdb, "protein", resid="GDP", operator="OR")
The argument verbose=TRUE can be used to print more information on how the selection components are combined:
sele <- atom.select(pdb, "protein", elety=c("N", "CA", "C"), resno=50:60, verbose=T)
##
## .. 00001340 atom(s) from 'string' selection
## .. 00000505 atom(s) from 'elety' selection
## .. 00000077 atom(s) from 'resno' selection
## .. 00000033 atom(s) in final combined selectionFunction combine.select() provides further functionality to combine selections. For example the above selection of protein and GDP can be completed with:
a.inds <- atom.select(pdb, "protein") b.inds <- atom.select(pdb, resid="GDP") sele <- combine.select(a.inds, b.inds, operator="OR")
See help(combine.select) for more information.
Question:
Using atom.select() how would you extract the amino acid sequence of your structure in 3-letter and 1-letter forms?
Answer:
First select the C-alpha atoms, and use the returned atom indices to access the resid values of pdb$atom.
ca.inds <- atom.select(pdb, "calpha") aa3 <- pdb$atom$resid[ ca.inds$atom ] head(aa3)
## [1] "MET" "THR" "GLU" "TYR" "LYS" "LEU"From there you can use the utility function aa321() to convert to 1-letter from
aa321(aa3)
## [1] "M" "T" "E" "Y" "K" "L" "V" "V" "V" "G" "A" "G" "G" "V" "G" "K" "S" "A"
## [19] "L" "T" "I" "Q" "L" "I" "Q" "N" "H" "F" "V" "D" "E" "Y" "D" "P" "T" "I"
## [37] "E" "D" "S" "Y" "R" "K" "Q" "V" "V" "I" "D" "G" "E" "T" "C" "L" "L" "D"
## [55] "I" "L" "D" "T" "A" "G" "Q" "E" "E" "Y" "S" "A" "M" "R" "D" "Q" "Y" "M"
## [73] "R" "T" "G" "E" "G" "F" "L" "C" "V" "F" "A" "I" "N" "N" "T" "K" "S" "F"
## [91] "E" "D" "I" "H" "Q" "Y" "R" "E" "Q" "I" "K" "R" "V" "K" "D" "S" "D" "D"
## [109] "V" "P" "M" "V" "L" "V" "G" "N" "K" "C" "D" "L" "A" "A" "R" "T" "V" "E"
## [127] "S" "R" "Q" "A" "Q" "D" "L" "A" "R" "S" "Y" "G" "I" "P" "Y" "I" "E" "T"
## [145] "S" "A" "K" "T" "R" "Q" "G" "V" "E" "D" "A" "F" "Y" "T" "L" "V" "R" "E"
## [163] "I" "R" "Q" "H" "K" "L"Note that if you try searching with help.search("PDB sequence", package="bio3d"), you will likely find a bio3d function that essentially does this all for you; Namely pdbseq():
## 1 2 3 4 5 6
## "M" "T" "E" "Y" "K" "L"Given the large number of functions in the bio3d package, using help.search() can be an effective way to find functionality related to your specific task.
Write a PDB object
Use the command write.pdb() to output a structure object to a PDB formatted file on your local hard drive. Below we use function atom.select() to select only the backbone atoms, and trim.pdb() to create a new PDB object based on our selection of backbone atoms. Finally we use the function write.pdb() to write the newly generated PDB file containing only the backbone atoms:
# Output a backbone only PDB file to disc b.inds <- atom.select(pdb, "back") backpdb <- trim.pdb(pdb, b.inds) write.pdb(backpdb, file="4q21_back.pdb")
Side-note:
The selection statement can directly be provided into function trim.pdb(). Alternatively, function atom.select() can also return the resulting trimmed pdb object using the optional argument value=TRUE. See examples below:
# Selection statements can be passed directly to trim.pdb() backpdb <- trim.pdb(pdb, "backbone") # The 'value=TRUE' option of atom.select() will result in a PDB object being returned backpdb <- atom.select(pdb, "backbone", value=TRUE)
Function write.pdb() contains further arguments to change the specific data in the PDB structure object. For example, using argument resno the residue numbers in the PDB object will be changed according to the input values, e.g. for the purpose of renumbering a PDB object (see also the force.renumber argument in clean.pdb() or the renumber argument in the convert.pdb() function):
Manipulate a PDB object
Basic functions for concatenating, trimming, splitting, converting, rotating, translating and superposing PDB files are available but often you will want to manipulate PDB objects in a custom way.
Below we provide a basic example of such a manipulation process where we read in a multi-chained PDB structure, reassign chain identifiers, and renumber selected residues.
pdb <- read.pdb("4lhy") # select chains A, E and F inds <- atom.select(pdb, chain=c("A", "E", "F")) # trim PDB to selection pdb2 <- trim.pdb(pdb, inds) # assign new chain identifiers pdb2$atom$chain[ pdb2$atom$chain=="E" ] <- "B" pdb2$atom$chain[ pdb2$atom$chain=="F" ] <- "C" # re-number chain B and C pdb2$atom$resno[ pdb2$atom$chain=="B" ] <- pdb2$atom$resno[ pdb2$atom$chain=="B" ] - 156 pdb2$atom$resno[ pdb2$atom$chain=="C" ] <- pdb2$atom$resno[ pdb2$atom$chain=="C" ] - 156 # assign the GDP residue a residue number of 500 pdb2$atom$resno[ pdb2$atom$resid=="GDP" ] <- 500 # use chain D for the GDP residue pdb2$atom$chain[ pdb2$atom$resid=="GDP" ] <- "D" # Center, to the coordinate origin, and orient, by principal axes, # the coordinates of a given PDB structure or xyz vector. xyz <- orient.pdb(pdb2) # write the new pdb object to file write.pdb(pdb2, xyz=xyz, file="4LHY_AEF-oriented.pdb")
Concatenate multiple PDBs
Function cat.pdb() can be used to concatenate two or more PDB files. This function contains further arguments to re-assign residue numbers and chain identifiers. In the example below we illustrate how to concatenate 4q21 with specific components of 4lhy into a new PDB object:
# read two G-protein structures a <- read.pdb("4q21") b <- read.pdb("4lhy") a1 <- trim.pdb(a, chain="A") b1 <- trim.pdb(b, chain="A") b2 <- trim.pdb(b, chain="E") b3 <- trim.pdb(b, chain="F") # concatenate PDBs new <- cat.pdb(a1, b1, b2, b3, rechain=TRUE) unique(new$atom$chain) # write new PDB object to file write.pdb(new, file="4Q21-4LHY.pdb")
Coordinate superposition and structural alignment
Structure superposition is often essential for the direct comparison of multiple structures. Bio3D offers versatile functionality for coordinate superposition at various levels. The simplest level is sequence only based superposition:
Here the returned object is of class pdbs, which we will discuss in detail further below. For now note that it contains a xyz numeric matrix of aligned C-alpha coordinates.
pdbs
## 1 . . . . 50
## [Truncated_Name:1]4q21.pdb MTEYKLVVVGAGGVGKSALTIQLIQNHFVDEYDPTIEDSYRKQVVIDGET
## [Truncated_Name:2]521p.pdb MTEYKLVVVGAVGVGKSALTIQLIQNHFVDEYDPTIEDSYRKQVVIDGET
## *********** **************************************
## 1 . . . . 50
##
## 51 . . . . 100
## [Truncated_Name:1]4q21.pdb CLLDILDTAGQEEYSAMRDQYMRTGEGFLCVFAINNTKSFEDIHQYREQI
## [Truncated_Name:2]521p.pdb CLLDILDTTGQEEYSAMRDQYMRTGEGFLCVFAINNTKSFEDIHQYREQI
## ******** *****************************************
## 51 . . . . 100
##
## 101 . . . . 150
## [Truncated_Name:1]4q21.pdb KRVKDSDDVPMVLVGNKCDLAARTVESRQAQDLARSYGIPYIETSAKTRQ
## [Truncated_Name:2]521p.pdb KRVKDSDDVPMVLVGNKCDLAARTVESRQAQDLARSYGIPYIETSAKTRQ
## **************************************************
## 101 . . . . 150
##
## 151 . 168
## [Truncated_Name:1]4q21.pdb GVEDAFYTLVREIRQHKL
## [Truncated_Name:2]521p.pdb GVEDAFYTLVREIRQH--
## ****************
## 151 . 168
##
## Call:
## pdbaln(files = c("4q21", "521p"), fit = TRUE)
##
## Class:
## pdbs, fasta
##
## Alignment dimensions:
## 2 sequence rows; 168 position columns (166 non-gap, 2 gap)
##
## + attr: xyz, resno, b, chain, id, ali, resid, sse, callUnderlying the pdbfit() function are calls to the seqaln() and fit.xyz() functions. The later of theses does the actual superposition based on all the aligned positions returned from seqaln(). Hence the superposition is said to be sequence based.
An alternative approach is to use a structure only alignment method (e.g. the mustang() function) as a basis for superposition. This is particularly useful when the structures (and hence usually the sequences) to be compared are dissimilar to the point where sequence comparison may give erroneous results.
Variations of both these approaches are also implemented in bio3d. For example, the function struct.aln() performs cycles of refinement steps of the alignment to improve the fit by removing atoms with a high structural deviation.
Note: The seqaln() function requires MUSCLE
The MUSCLE multiple sequence alignment program (available from the muscle home page) must be installed on your system and in the search path for executables in order to run functions pdbfit() and struct.aln() as these call the seqaln() function, which is based on MUSCLE. Please see the installation vignette for further details.
Function struct.aln() performs a sequence alignment followed by a structural alignment of two PDB objects. This facilitates rapid superposition of two PDB structures with unequal, but related PDB sequences. Below we use struct.aln() to superimpose the multi-chained PDB ID 4lhy to PDB ID 4q21:
# read two G-protein structures a <- read.pdb("4q21") b <- read.pdb("4lhy") # perform iterative alignment aln <- struct.aln(a, b) # store new coordinates of protein B b$xyz <- aln$xyz
Note that struct.aln() performs cycles of refinement steps of the structural alignment to improve the fit by removing atoms with a high structural deviation. At each cycle it prints associated RMSD value of atoms included in the alignment. The resulting superimposed structures are optionally written to your hard drive at default folder with name fitlsq/.

Result of fitting PDB IDs 4lhy (grey) and 4q21 (red) using function struct.aln(). Note that 4lhy contains 6 chains (A-F), while 4q21 consist of only 1 chain. Based on the sequence and structure alignment performed internally in struct.aln() the two PDBs align at certain helices in the shared domain.
While struct.aln() operates on a pair of PDB objects, function fit.xyz() can perform coordinate superposition on multiple structures. However, indices needs to provided ensuring the same number of atoms for which the fitting should be based. Below we superimpose a specific helix of 4lhy onto an equivalent helix of 4q21:
# indices at which the superposition should be based a.ind <- atom.select(a, chain="A", resno=87:103, elety="CA") b.ind <- atom.select(b, chain="A", resno=93:109, elety="CA") # perform superposition xyz <- fit.xyz(fixed=a$xyz, mobile=b$xyz, fixed.inds=a.ind$xyz, mobile.inds=b.ind$xyz) # write coordinates to file write.pdb(b, xyz=xyz, file="4LHY-at-4Q21.pdb")
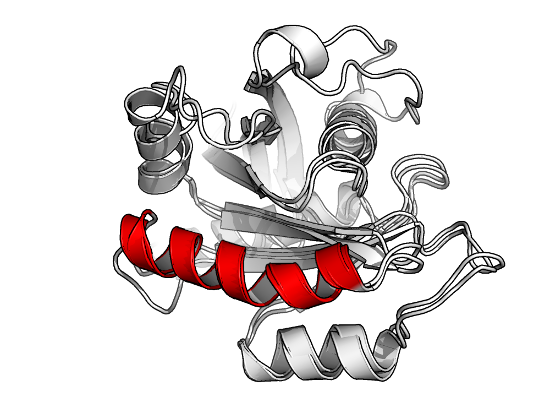
Result of fitting a specific helix in PDB ID 4lhy onto an equivalent helix of PDB ID 4q21 using function fit.xyz().
For more information on Coordinate superposition see help(fit.xyz).
Binding site identification
Function binding.site() provides functionality to determine interacting residues between two PDB enteties or between two atom selections of a PDB object. This function reports the residues of selection 1 closer than a cutoff to selection 2. By default, binding.site() attempts to identify “protein” and “ligand” using atom.select(). Thus, to determine the binding site of PDB 4q21 only the pdb object needs to be provided:
# read G-protein structure pdb <- read.pdb("4q21") bs <- binding.site(pdb)
The output of binding.site() is a list containing (1) an inds component with a select object containing the atom and xyz indices of the identified binding site, (2) a resnames component containing formatted residue names of the binding site residues, (3) resno and chain components with residue numbers and chain identifiers for the binding site residues.
# residue names of identified binding site print(bs$resnames)
## [1] "ALA 11 (A)" "GLY 12 (A)" "GLY 13 (A)" "VAL 14 (A)" "GLY 15 (A)"
## [6] "LYS 16 (A)" "SER 17 (A)" "ALA 18 (A)" "PHE 28 (A)" "VAL 29 (A)"
## [11] "ASP 30 (A)" "GLU 31 (A)" "TYR 32 (A)" "ASP 33 (A)" "PRO 34 (A)"
## [16] "ILE 36 (A)" "ASP 57 (A)" "THR 58 (A)" "ALA 59 (A)" "ASN 116 (A)"
## [21] "LYS 117 (A)" "ASP 119 (A)" "LEU 120 (A)" "THR 144 (A)" "SER 145 (A)"
## [26] "ALA 146 (A)" "LYS 147 (A)"Alternatively, user defined atom selections can be provided to determine e.g. residues at the binding interface between two proteins:
b <- read.pdb("4lhy") # atom selection a.inds <- atom.select(b, chain="A") b.inds <- atom.select(b, chain=c("E", "F")) # identify interface residues bs <- binding.site(b, a.inds=a.inds, b.inds=b.inds) # use b-factor column to store interface in PDB file b$atom$b[ bs$inds$atom ] <- 1 b$atom$b[ -bs$inds$atom ] <- 0 # write to file write.pdb(b, file="4LHY-interface.pdb")

Visualization of interface residues as obtained from function binding.site().
Reading multi-model PDB files
Bio3D can read and analyze multi-model PDB files, such as those representing NMR ensembles. To enable reading of all models you need to provide argument multi=TRUE to function read.pdb(). This will store the Cartesian coordinates of each model in the xyz component so that nrow(xyz) equals the number of models in the PDB structure. Several Bio3D functions works directly on these multi-model PDB objects. Below we provide an example of how to read and access the coordinates of a multi-model PDB file. We then illustrate two functions for the analysis of a multi-model PDB file.
# Read multi-model PDB file pdb <- read.pdb("1d1d", multi=TRUE)
## Note: Accessing on-line PDB file# The xyz component contains 20 frames pdb$xyz
##
## Total Frames#: 20
## Total XYZs#: 10185, (Atoms#: 3395)
##
## [1] 1.325 0 0 <...> -16.888 -81.895 5.777 [203700]
##
## + attr: Matrix DIM = 20 x 10185# Select a subset of the protein ca.inds <- atom.select(pdb, "calpha") # Access C-alpha coordinates of the first 5 models #pdb$xyz[1:5, ca.inds$xyz]
Identification of dynamic domains
Function geostas() from the Bio3D package, bio3d.geostas, attempts to identify rigid domains in a protein (or nucleic acid) structure based on knowledge of its structural ensemble (as obtained a from multi-model PDB input file, an ensemble of multiple PDB files (see section below), NMA or MD results). Below we demonstrate the identification of such dynamic domains in a multi-model PDB and use function fit.xyz() to fit all models to one of the identified domains. We then use write.pdb() to store the aligned structures for further visualization, e.g. in VMD:
library(bio3d.geostas)
## Loading required package: bio3d.nma## Registered S3 methods overwritten by 'bio3d.nma':
## method from
## dccm.egnm bio3d
## dccm.enma bio3d
## dccm.gnm bio3d
## dccm.nma bio3d
## mktrj.enma bio3d
## mktrj.nma bio3d
## plot.enma bio3d
## plot.nma bio3d
## plot.rmsip bio3d
## print.enma bio3d
## print.nma bio3d
## pymol.nma bio3d##
## Attaching package: 'bio3d.nma'## The following objects are masked from 'package:bio3d':
##
## aanma, bhattacharyya, build.hessian, cov.enma, cov.nma,
## covsoverlap, difference.vector, ff.aaenm, ff.aaenm2, ff.anm,
## ff.calpha, ff.pfanm, ff.reach, ff.sdenm, fluct.nma, gnm,
## load.enmff, nma, overlap, rmsip, rtb, sip, var.pdbs, var.xyz## Registered S3 method overwritten by 'bio3d.geostas':
## method from
## plot.geostas bio3d##
## Attaching package: 'bio3d.geostas'## The following objects are masked from 'package:bio3d':
##
## amsm.xyz, geostas# Domain analysis gs <- geostas(pdb)
## .. 220 'calpha' atoms selected
## .. 'xyz' coordinate data with 20 frames
## .. 'fit=TRUE': running function 'core.find'
## .. coordinates are superimposed to core region
## .. calculating atomic movement similarity matrix ('amsm.xyz()')
## .. dimensions of AMSM are 220x220
## .. clustering AMSM using 'kmeans'
## .. converting indices to match input 'pdb' object
## (additional attribute 'atomgrps' generated)# Fit all frames to the 'first' domain domain.inds <- gs$inds[[1]] xyz <- pdbfit(pdb, inds=domain.inds) # write fitted coordinates write.pdb(pdb, xyz=xyz, chain=gs$atomgrps, file="1d1d_fit-domain1.pdb") # plot geostas results plot(gs, contour=FALSE)
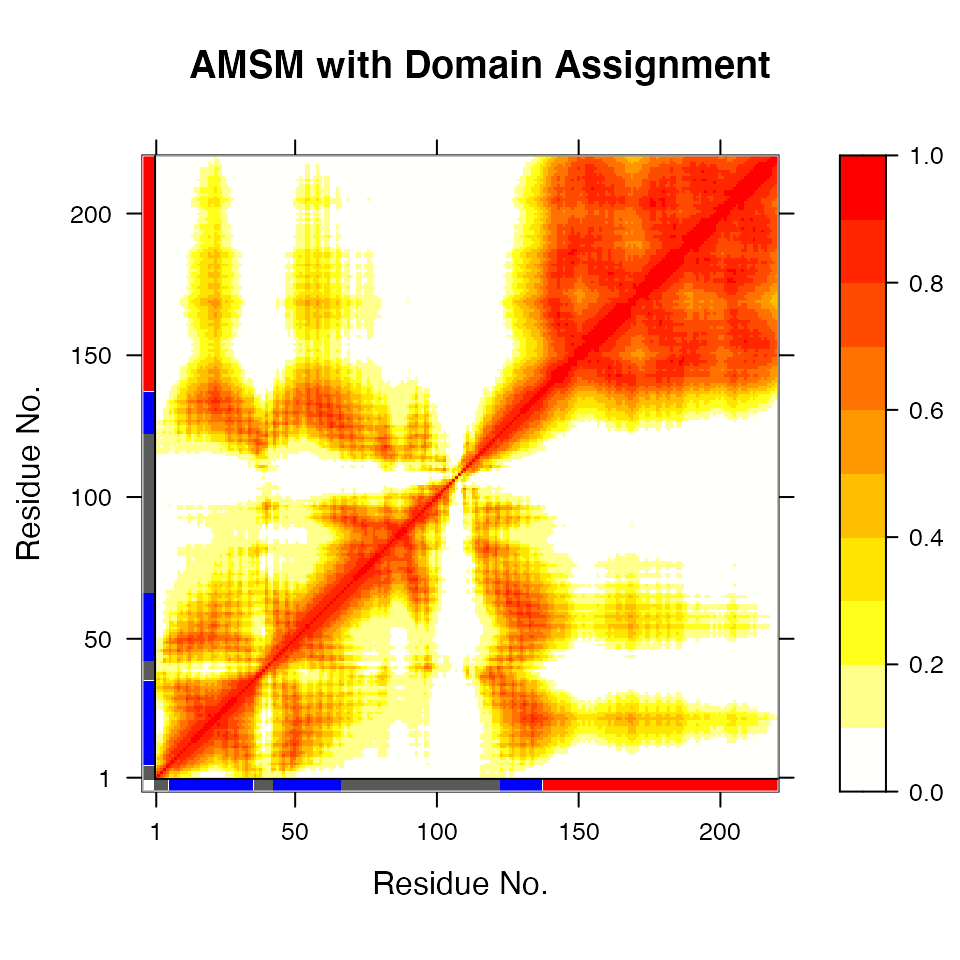
Plot of atomic movement similarity matrix with domain annotation for PDB ID 1d1d

Multi-model PDB with 20 frames superimposed on ‘domain 1’ (colored grey) identified using function geostas(). See also related function core.find().
Invariant core identification
Function core.find() also works on a multi-model PDB file (or pdbs object, see below) to determine the most invariant region a protein ensemble. Below we fit the NMR ensemble to the region identified by core.find():
Constructing biological units
PDB crystal structure files typically detail the atomic coordinates for the contents of just one crystal asymmetric unit. This may or may not be the same as the biologically-relevant assembly or biological unit. For example, the PDB entry ‘2DN1’ of human hemoglobin stores one pair of alpha and beta globin subunits (see Figure 7). However, under physiological conditions the biological unit is known to be a tetramer (composed of two alpha and two beta subunits). In this particular case, the crystal asymmetric unit is part of the biological unit. More generally, the asymmetric unit stored in a PDB file can be: (i) One biological unit; (ii) A portion of a biological unit, or (iii) Multiple biological units.
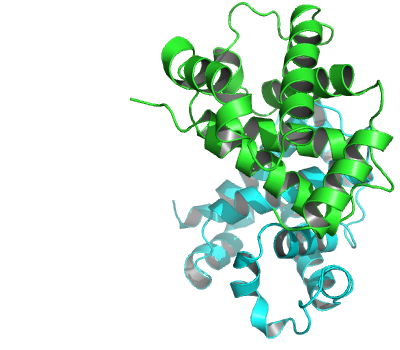
The alpha/beta dimer of human hemoglobin as stored in the PDB entry ‘2DN1’.
Reconstruction of the biological unit for a given PDB file can thus be essential for understanding structure-function relationships. PDB files often provide information about how to construct biological units via symmetry operations (i.e. rotation and translation) performed on the stored coordinates. In bio3d, the function read.pdb() automatically stores this information if available. The function biounit() can then be called to build biological units based on this information stored within an appropriate ‘pdb’ object. The following example illustrates the construction of the hemoglobin tetramer from the dimer in the asymmetric unit of ‘2DN1’.
# Read PDB from online database pdb <- read.pdb("2dn1")
## Note: Accessing on-line PDB file# Examine biological unit matrices pdb$remark$biomat
## $num
## [1] 1
##
## $chain
## $chain[[1]]
## [1] "A" "B"
##
##
## $mat
## $mat[[1]]
## $mat[[1]]$`A B`
## [,1] [,2] [,3] [,4]
## [1,] 1 0 0 0
## [2,] 0 1 0 0
## [3,] 0 0 1 0
##
## $mat[[1]]$`A B`
## [,1] [,2] [,3] [,4]
## [1,] 0 1 0 0
## [2,] 1 0 0 0
## [3,] 0 0 -1 0
##
##
##
## $method
## [1] "AUTHOR"The $remark$biomat component in a ‘pdb’ object describe the transformation (translation and rotation) matrices that can be applied to construct biological units. It contains:
- num, the number of biological units described
- chain, a ‘list’ object. Each component is a vector of chain labels to which the transformation matrices are applied to get the corresponding biological unit
- mat, a ‘list’ of transformation matrices for each biological unit
- method, a character vector about the method (‘AUTHOR’ or ‘SOFTWARE’) that determined each biological unit
biounit(pdb)
## $`AUTHOR.determined.tetramer (4 chains)`
##
## Call: biounit(pdb = pdb)
##
## Total Models#: 1
## Total Atoms#: 5012, XYZs#: 15036 Chains#: 4 (values: A B C D)
##
## Protein Atoms#: 4344 (residues/Calpha atoms#: 570)
## Nucleic acid Atoms#: 0 (residues/phosphate atoms#: 0)
##
## Non-protein/nucleic Atoms#: 668 (residues: 472)
## Non-protein/nucleic resid values: [ HEM (4), HOH (460), MBN (4), OXY (4) ]
##
## Protein sequence:
## LSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHGKK
## VADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAV
## HASLDKFLASVSTVLTSKYRHLTPEEKSAVTALWGKVNVDEVGGEALGRLLVVYPWTQRF
## FESFGDLSTPDAVMGNPKVKAHGKKVLGAFSDGLAHLDNLKGTFA...<cut>...HKYH
##
## + attr: atom, helix, sheet, seqres, xyz,
## calpha, call, logThe function biounit() returns a ‘list’ object. Each component of the ‘list’ represents a possible biological unit stored as a ‘pdb’ object. In above example, two new chains (chain C and D) are constructed from symmetry operations performed on the original chains (chain A and B) see Figure 8.
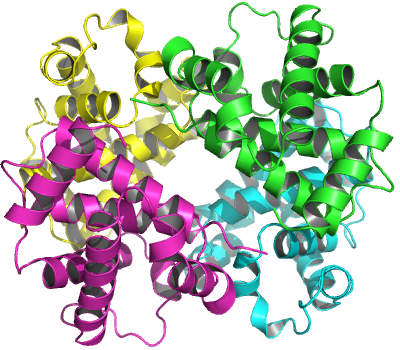
The biological unit of human hemoglobin (tetramer) built from PDB entry ‘2DN1’.
Side-note:
The biological unit described in a PDB file is not always unique. Distinct methods, e.g. based on expertise of authors of the crystallographic structure or calucaltion of software (See here for more details), may determine different multimeric state. Also, as mentioned above, one crystal asymmetric unit may contain multiple biological units. The function biounit() returns all the possible biological units listed in the file (and that is why the return value is a ‘list’ instead of a simple ‘pdb’ object). It is dependent on users which biological unit is finally adopted for further analysis. Check the ‘names’ attributes of the returned ‘list’ for the determination method and multimeric state of each biological unit. For example, the PDB entry ‘1KJY’ describes three possible biological units for the G protein alpha subunit bound with GDI inhibitor. The common way is to select either one of the two dimers (they have slightly distinct conformation) instead of the software predicted tetramer.
pdb <- read.pdb("1kjy")
## Note: Accessing on-line PDB file## [1] "AUTHOR.determined.dimer (2 chains)"
## [2] "AUTHOR.determined.dimer (2 chains)"
## [3] "SOFTWARE.determined.tetramer (4 chains)"Optionally, biounit() returns the biological unit as a multi-model ‘pdb’ object. In this case, the topology of the ‘pdb’ (e.g. $atom) is the same as input, whereas additional coordinates generated via the symmetry operations are stored in distinc rows of $xyz. This is obtained by setting the argument multi=TRUE. It is particular useful for speed enhancements with biological units containing many symmetric copies, for example a virus capsid:
pdb <- read.pdb("2bfu")
## Note: Accessing on-line PDB filebio <- biounit(pdb, multi = TRUE) bio
## $`SOFTWARE.determined.multimer (120 chains)`
##
## Call: biounit(pdb = pdb, multi = TRUE)
##
## Total Models#: 60
## Total Atoms#: 5282, XYZs#: 950760 Chains#: 2 (values: L S)
##
## Protein Atoms#: 5282 (residues/Calpha atoms#: 558)
## Nucleic acid Atoms#: 0 (residues/phosphate atoms#: 0)
##
## Non-protein/nucleic Atoms#: 0 (residues: 0)
## Non-protein/nucleic resid values: [ none ]
##
## Protein sequence:
## MEQNLFALSLDDTSSVRGSLLDTKFAQTRVLLSKAMAGGDVLLDEYLYDVVNGQDFRATV
## AFLRTHVITGKIKVTATTNISDNSGCCLMLAINSGVRGKYSTDVYTICSQDSMTWNPGCK
## KNFSFTFNPNPCGDSWSAEMISRSRVRMTVICVSGWTLSPTTDVIAKLDWSIVNEKCEPT
## IYHLADCQNWLPLNRWMGKLTFPQGVTSEVRRMPLSIGGGAGATQ...<cut>...TPPL
##
## + attr: atom, helix, sheet, seqres, xyz,
## calpha, callbio[[1]]$xyz
##
## Total Frames#: 60
## Total XYZs#: 15846, (Atoms#: 5282)
##
## [1] 21.615 -8.176 108.9 <...> -109.546 -20.972 -91.006 [950760]
##
## + attr: Matrix DIM = 60 x 15846Working with multiple PDB files
The Bio3D packages were designed to specifically facilitate the analysis of multiple structures from both experiment and simulation. The challenge of working with these structures is that they are usually different in their composition (i.e. contain differing number of atoms, sequences, chains, ligands, structures, conformations etc. even for the same protein as we will see below) and it is these differences that are frequently of most interest.
For this reason Bio3D contains extensive utilities to enable the reading, writing, manipulation and analysis of such heterogenous structure sets. This topic is detailed extensively in the separate Principal Component Analysis vignette available from http://thegrantlab.org/bio3d.
Before delving into more advanced analysis (detailed in additional vignettes) lets examine how we can read multiple PDB structures from the RCSB PDB for a particular protein and perform some basic analysis:
# Download some example PDB files ids <- c("1TND_B","1AGR_A","1FQJ_A","1TAG_A","1GG2_A","1KJY_A") raw.files <- get.pdb(ids)
The get.pdb() function will download the requested files, below we extract the particular chains we are most interested in with the function pdbsplit() (note these ids could come from the results of a blast.pdb() search as described in other vignettes). The requested chains are then aligned and their structural data stored in a new object pdbs that can be used for further analysis and manipulation.
# Extract and align the chains we are interested in files <- pdbsplit(raw.files, ids) pdbs <- pdbaln(files)
Below we examine the sequence and structural similarity.
# Calculate sequence identity pdbs$id <- basename.pdb(pdbs$id) seqidentity(pdbs)
## 1TND_B 1AGR_A 1FQJ_A 1TAG_A 1GG2_A 1KJY_A
## 1TND_B 1.000 0.693 0.914 1.000 0.690 0.696
## 1AGR_A 0.693 1.000 0.779 0.694 0.997 0.994
## 1FQJ_A 0.914 0.779 1.000 0.914 0.776 0.782
## 1TAG_A 1.000 0.694 0.914 1.000 0.691 0.697
## 1GG2_A 0.690 0.997 0.776 0.691 1.000 0.991
## 1KJY_A 0.696 0.994 0.782 0.697 0.991 1.000## Calculate RMSD rmsd(pdbs, fit=TRUE)
## 1TND_B 1AGR_A 1FQJ_A 1TAG_A 1GG2_A 1KJY_A
## 1TND_B 0.000 0.965 0.609 1.283 1.612 2.100
## 1AGR_A 0.965 0.000 0.873 1.575 1.777 1.914
## 1FQJ_A 0.609 0.873 0.000 1.265 1.737 2.042
## 1TAG_A 1.283 1.575 1.265 0.000 1.687 1.841
## 1GG2_A 1.612 1.777 1.737 1.687 0.000 1.879
## 1KJY_A 2.100 1.914 2.042 1.841 1.879 0.000
Results of protein structure principal component analysis (PCA) plotted by function plot.pca()
Below we examine protein structural dynamics predicted by normal mode analysis using the bio3d.nma package from the Bio3D package family.3
## Quick NMA of all structures (see Figure 10) library(bio3d.nma) modes <- nma(pdbs) plot(modes, pdbs, spread=TRUE)
## Extracting SSE from pdbs$sse attribute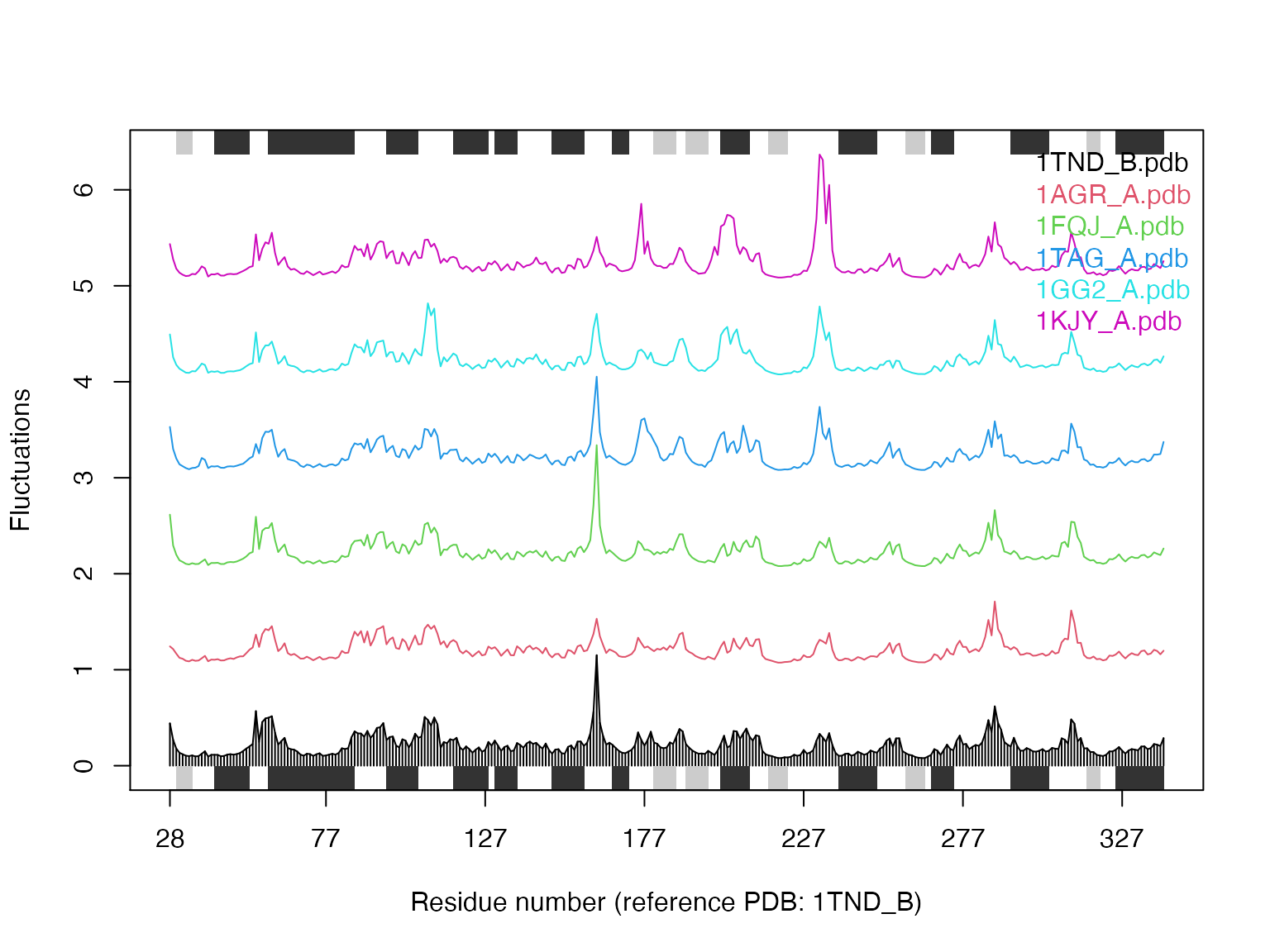
Results of ensemble normal mode analysis (NMA) plotted by function plot.enma()
Question:
What effect does setting the fit=TRUE option have in the RMSD calculation? What would the results indicate if you set fit=FALSE or disparaged this option? HINT: Bio3D functions have various default options that will be used if the option is not explicitly specified by the user, see help(rmsd) for an example and note that the input options with an equals sign (e.g. fit=FALSE) have default values.
Where to next
If you have read this far, congratulations! We are ready to have some fun and move to other package vignettes that describe more interesting analysis including advanced Comparative Structure Analysis (where we will mine available experimental data and supplement it with simulation results to map the conformational dynamics and coupled motions of proteins), Trajectory Analysis (where we analyze molecular dynamics simulation trajectories), enhanced methods for Normal Mode Analysis (where we will explore the dynamics of large protein families and superfamilies), and Correlation Network Analysis (where we will build and dissect dynamic networks form different correlated motion data).
Document and current Bio3D session details
This document is available on the Bio3D website in both HTML and PDF formats. All code can be extracted and automatically executed to generate Figures and/or the PDF/HTML with the following commands:
# Information about the current Bio3D session sessionInfo()
## R version 4.0.2 (2020-06-22)
## Platform: x86_64-apple-darwin17.0 (64-bit)
## Running under: macOS Catalina 10.15.6
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRblas.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] bio3d.geostas_0.1.0.9000 bio3d.nma_0.1.0.9000 bio3d_2.4-1.9000
##
## loaded via a namespace (and not attached):
## [1] Rcpp_1.0.5 cpp11_0.2.1 knitr_1.29 magrittr_1.5
## [5] lattice_0.20-41 R6_2.4.1 ragg_0.3.1 rlang_0.4.7
## [9] stringr_1.4.0 highr_0.8 tools_4.0.2 parallel_4.0.2
## [13] grid_4.0.2 xfun_0.17 htmltools_0.5.0 systemfonts_0.3.1
## [17] yaml_2.2.1 assertthat_0.2.1 rprojroot_1.3-2 digest_0.6.25
## [21] pkgdown_1.6.1.9000 crayon_1.3.4 fs_1.5.0 memoise_1.1.0
## [25] evaluate_0.14 rmarkdown_2.3 stringi_1.5.3 compiler_4.0.2
## [29] desc_1.2.0 backports_1.1.9References
Grant, B. J., A. P. D. C Rodrigues, K. M. Elsawy, A. J. Mccammon, and L. S. D. Caves. 2006. “Bio3d: An R Package for the Comparative Analysis of Protein Structures.” Bioinformatics 22: 2695–6. https://doi.org/10.1093/bioinformatics/btl461.
Skjaerven, L., X. Q. Yao, G. Scarabelli, and B. J. Grant. 2014. “Integrating Protein Structural Dynamics and Evolutionary Analysis with Bio3d.” BMC Bioinformatics 15: 399. https://doi.org/10.1186/s12859-014-0399-6.
The latest version of the package, full documentation and further vignettes (including detailed installation instructions) can be obtained from the main Bio3D website: thegrantlab.org/bio3d/.↩︎
This vignette contains executable examples, see
help(vignette)for further details.↩︎See also dedicated vignettes for ensemble NMA provided with the Bio3D package.↩︎