Ensemble Normal Mode Analysis with All-Atom ENM
aanma.pdbs.RdPerform normal mode analysis (NMA) on an ensemble of aligned protein structures using all-atom elastic network model (aaENM).
# S3 method for pdbs aanma(pdbs, fit = TRUE, full = FALSE, subspace = NULL, rm.gaps = TRUE, ligand = FALSE, outpath = NULL, gc.first = TRUE, ncore = NULL, ...)
Arguments
| pdbs | an ‘pdbs’ object as obtained from |
|---|---|
| fit | logical, if TRUE C-alpha coordinate based superposition is performed prior to normal mode calculations. |
| full | logical, if TRUE return the complete, full structure, ‘nma’ objects. |
| subspace | number of eigenvectors to store for further analysis. |
| rm.gaps | logical, if TRUE obtain the hessian matrices for only atoms in the aligned positions (non-gap positions in all aligned structures). Thus, gap positions are removed from output. |
| ligand | logical, if TRUE ligand molecules are also included in the calculation. |
| outpath | character string specifing the output directory to which the PDB structures should be written. |
| gc.first | logical, if TRUE will call gc() first before mode calculation
for each structure. This is to avoid memory overload when
|
| ncore | number of CPU cores used to do the calculation. |
| ... | additional arguments to |
Value
Returns a list of ‘nma’ objects (outmodes is provided
and is not ‘calpha’) or an ‘enma’ object with the following
components:
a numeric matrix containing aligned atomic fluctuations with one row per input structure.
a numeric matrix of pair wise RMSIP values (only the ten lowest frequency modes are included in the calculation).
a three-dimensional array with aligned eigenvectors (corresponding to the subspace defined by the first N non-trivial eigenvectors (‘U’) of the ‘nma’ object).
numeric matrix containing the raw eigenvalues with one row per input structure.
a list with a nma object for each input
structure (available only when full=TRUE).
Details
This function builds elastic network model (ENM) using all heavy
atoms and performs subsequent normal mode analysis (NMA) on a set of
aligned protein structures obtained with function read.all.
The main purpose is to automate ensemble normal mode analysis using
all-atom ENMs.
By default, the effective Hessian for all C-alpha atoms is calculated
based on the Hessian built from all heavy atoms (including ligand atoms if
ligand=TRUE). Returned values include aligned mode vectors and
(when full=TRUE) a list containing the full ‘nma’ objects
one per each structure. When ‘rm.gaps=TRUE’ the unaligned atoms
are ommited from output. With default arguments ‘rmsip’ provides
RMSIP values for all pairwise structures.
When outmodes is provided and is not ‘calpha’
(e.g. ‘noh’. See aanma for more details), the
function simply returns a list of ‘nma’ objects, one per each
structure, and no aligned mode vector is returned. In this case, the
arguments full, subspace, and rm.gaps are ignored.
This is equivalent to a wrapper function repeatedly calling
aanma.
Author
Xin-Qiu Yao & Lars Skjaerven
See also
For normal mode analysis on single structure PDB:
aanma
For conventional C-alpha based normal mode analysis:
nma, nma.pdbs.
For the analysis of the resulting ‘eNMA’ object:
mktrj.enma, dccm.enma,
plot.enma, cov.enma.
Similarity measures:
sip, covsoverlap,
bhattacharyya, rmsip.
Related functionality:
read.all.
Examples
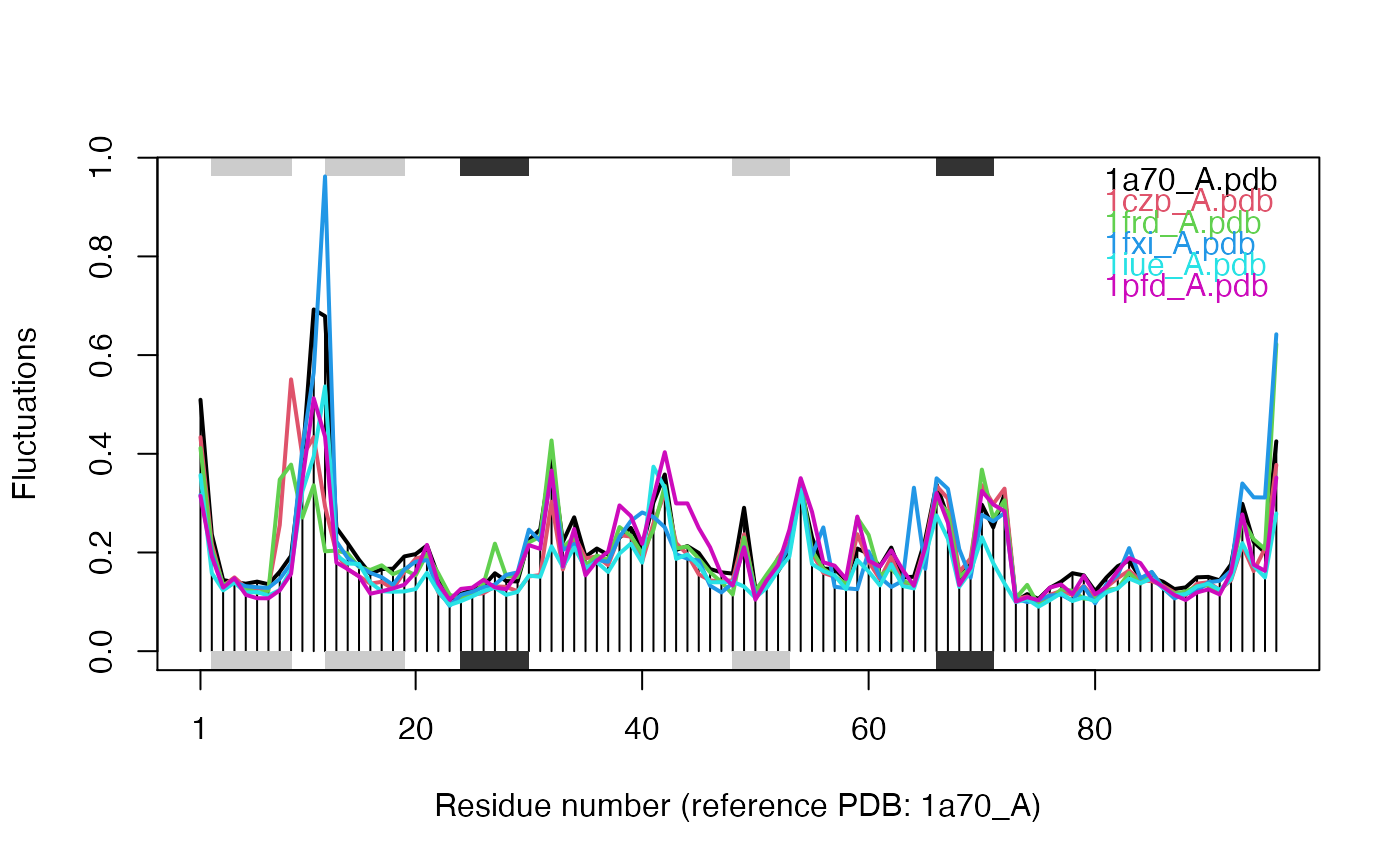
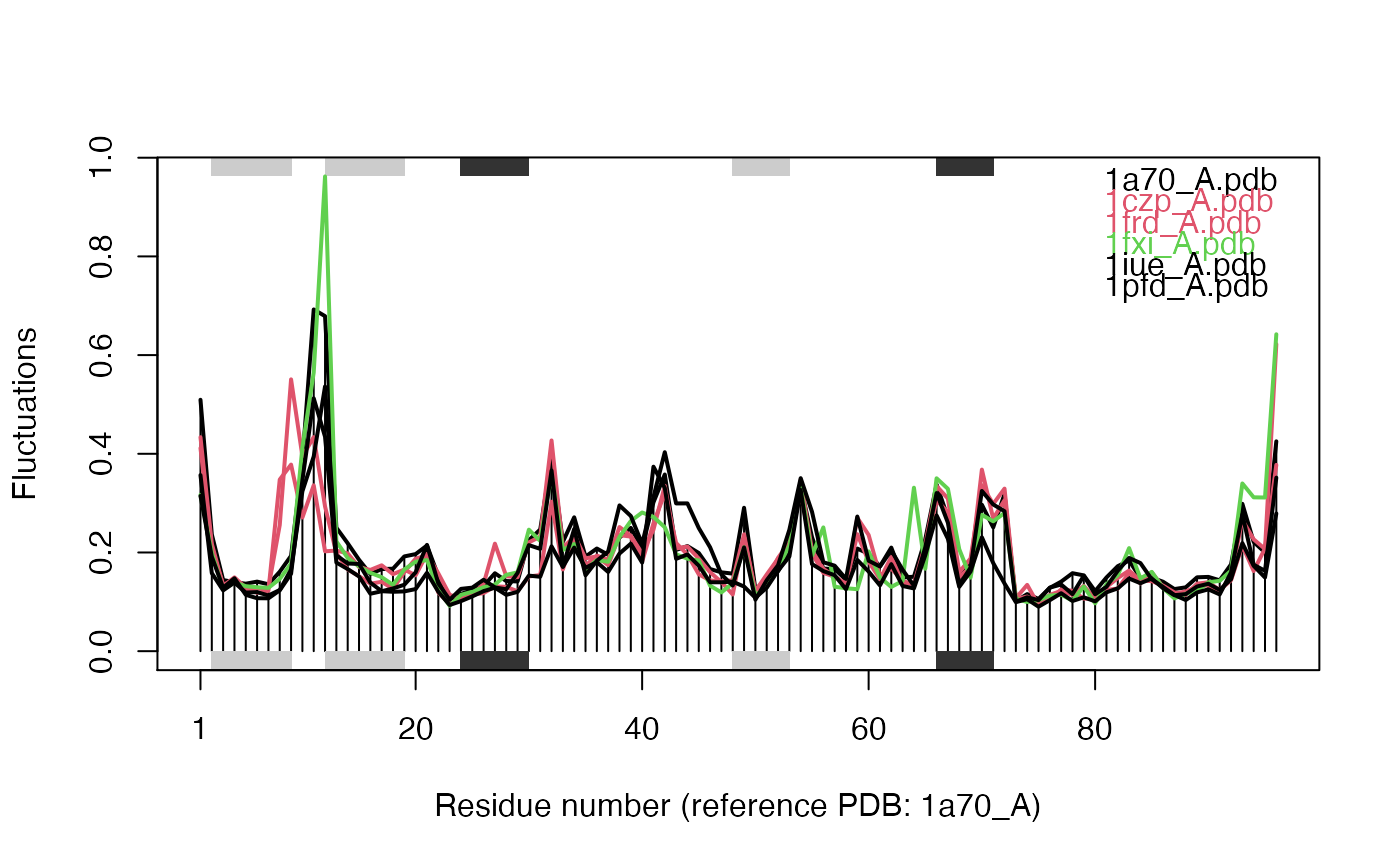
# \donttest{ # Needs MUSCLE installed - testing excluded if(check.utility("muscle")) { ## Fetch PDB files and split to chain A only PDB files ids <- c("1a70_A", "1czp_A", "1frd_A", "1fxi_A", "1iue_A", "1pfd_A") files <- get.pdb(ids, split = TRUE, path = tempdir()) ## Sequence Alignement aln <- pdbaln(files, outfile = tempfile()) ## Read all pdb coordinates pdbs <- read.all(aln) ## Normal mode analysis on aligned data modes <- aanma(pdbs, rm.gaps=TRUE) ## Plot fluctuation data plot(modes, pdbs=pdbs) ## Cluster on Fluctuation similariy sip <- sip(modes) hc <- hclust(dist(sip)) col <- cutree(hc, k=3) ## Plot fluctuation data plot(modes, pdbs=pdbs, col=col) ## RMSIP is pre-calculated heatmap(1-modes$rmsip) ## Bhattacharyya coefficient bc <- bhattacharyya(modes) heatmap(1-bc) }#> | | | 0% | |============ | 17% | |======================= | 33% | |=================================== | 50% | |=============================================== | 67% | |========================================================== | 83% | |======================================================================| 100% #> Reading PDB files: #> /var/folders/xf/qznxnpf91vb1wm4xwgnbt0xr0000gn/T//Rtmp9oBdbc/split_chain/1a70_A.pdb #> /var/folders/xf/qznxnpf91vb1wm4xwgnbt0xr0000gn/T//Rtmp9oBdbc/split_chain/1czp_A.pdb #> /var/folders/xf/qznxnpf91vb1wm4xwgnbt0xr0000gn/T//Rtmp9oBdbc/split_chain/1frd_A.pdb #> /var/folders/xf/qznxnpf91vb1wm4xwgnbt0xr0000gn/T//Rtmp9oBdbc/split_chain/1fxi_A.pdb #> /var/folders/xf/qznxnpf91vb1wm4xwgnbt0xr0000gn/T//Rtmp9oBdbc/split_chain/1iue_A.pdb #> /var/folders/xf/qznxnpf91vb1wm4xwgnbt0xr0000gn/T//Rtmp9oBdbc/split_chain/1pfd_A.pdb #> . PDB has ALT records, taking A only, rm.alt=TRUE #> ..... #> #> Extracting sequences #> #> pdb/seq: 1 name: /var/folders/xf/qznxnpf91vb1wm4xwgnbt0xr0000gn/T//Rtmp9oBdbc/split_chain/1a70_A.pdb #> pdb/seq: 2 name: /var/folders/xf/qznxnpf91vb1wm4xwgnbt0xr0000gn/T//Rtmp9oBdbc/split_chain/1czp_A.pdb #> PDB has ALT records, taking A only, rm.alt=TRUE #> pdb/seq: 3 name: /var/folders/xf/qznxnpf91vb1wm4xwgnbt0xr0000gn/T//Rtmp9oBdbc/split_chain/1frd_A.pdb #> pdb/seq: 4 name: /var/folders/xf/qznxnpf91vb1wm4xwgnbt0xr0000gn/T//Rtmp9oBdbc/split_chain/1fxi_A.pdb #> pdb/seq: 5 name: /var/folders/xf/qznxnpf91vb1wm4xwgnbt0xr0000gn/T//Rtmp9oBdbc/split_chain/1iue_A.pdb #> pdb/seq: 6 name: /var/folders/xf/qznxnpf91vb1wm4xwgnbt0xr0000gn/T//Rtmp9oBdbc/split_chain/1pfd_A.pdb #> Fitting pdb structuresdone #> #> Details of Scheduled Calculation: #> ... 6 input structures #> ... storing 282 eigenvectors for each structure #> ... dimension of x$U.subspace: ( 288x282x6 ) #> ... coordinate superposition prior to NM calculation #> ... aligned eigenvectors (gap containing positions removed) #> ... estimated memory usage of final 'eNMA' object: 3.7 Mb #>#>#>#> Calculating covariance matrices #> Calculating pairwise bhattacharyya coefs# }