Filter for Cross-correlation Matrices (Cij)
Usage
filter.dccm(x, cutoff.cij = 0.4, cmap = NULL, xyz = NULL, fac = NULL, cutoff.sims = NULL, collapse = TRUE, extra.filter = NULL, ...)
Arguments
- x
- A matrix (nXn), a numeric array with 3 dimensions (nXnXm), a list with m cells each containing nXn matrix, or a list with ‘all.dccm’ component, containing atomic correlation values, where "n" is the number of residues and "m" the number of calculations. The matrix elements should be in between -1 and 1. See ‘dccm’ function in bio3d package for further details.
- cutoff.cij
- Threshold for each individual correlation value. See below for details.
- cmap
- logical or numerical matrix indicating the contact map.
If logical and TRUE, contact map will be calculated with input
xyz. - xyz
- XYZ coordinates for distance matrix calculation.
- fac
- factor indicating distinct categories of input correlation matrices.
- cutoff.sims
- Threshold for the number of simulations with observed correlation
value above
cutoff.cijfor the same residue/atomic pairs. See below for details. - collapse
- logical, if TRUE the mean matrix will be returned.
- extra.filter
- Filter to apply in addition to the model chosen.
- ...
- extra arguments passed to function
cmap.
Description
This function builds various cij matrix for correlation network analysis
Value
-
Returns a matrix of class "dccm" or a 3D array of filtered cross-correlations.
References
Grant, B.J. et al. (2006) Bioinformatics 22, 2695--2696.
Details
If cmap is TRUE or provided a numerical matrix, the function inspects a set of cross-correlation matrices, or DCCM, and decides edges for correlation network analysis based on:
1. min(abs(cij)) >= cutoff.cij, or
2. max(abs(cij)) >= cutoff.cij && residues contact each other
based on results from cmap.
Otherwise, the function filters DCCMs with cutoff.cij and
return the mean of correlations present in at least
cutoff.sims calculated matrices.
Examples
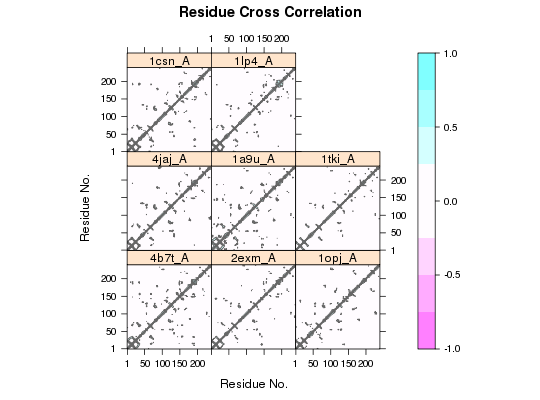
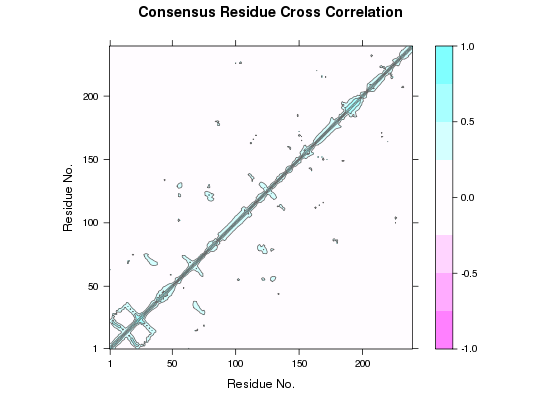
# Example of transducin attach(transducin) gaps.pos <- gap.inspect(pdbs$xyz) modes <- nma.pdbs(pdbs, ncore=NULL)Warning message: 3QI2_B might have missing residue(s) in structure: Fluctuations at neighboring positions may be affected.Details of Scheduled Calculation: ... 53 input structures ... storing 909 eigenvectors for each structure ... dimension of x$U.subspace: ( 915x909x53 ) ... coordinate superposition prior to NM calculation ... aligned eigenvectors (gap containing positions removed) ... estimated memory usage of final 'eNMA' object: 336.8 Mb | | | 0%dccms <- dccm.enma(modes, ncore=NULL) cij <- filter.dccm(dccms, xyz=pdbs)|======================================================================| 100%# Example protein kinase # Select Protein Kinase PDB IDs ids <- c("4b7t_A", "2exm_A", "1opj_A", "4jaj_A", "1a9u_A", "1tki_A", "1csn_A", "1lp4_A") # Download and split by chain ID files <- get.pdb(ids, path = "raw_pdbs", split=TRUE)|======================================================================| 100%# Alignment of structures pdbs <- pdbaln(files) # Sequence identityReading PDB files: raw_pdbs/split_chain/4b7t_A.pdb raw_pdbs/split_chain/2exm_A.pdb raw_pdbs/split_chain/1opj_A.pdb raw_pdbs/split_chain/4jaj_A.pdb raw_pdbs/split_chain/1a9u_A.pdb raw_pdbs/split_chain/1tki_A.pdb raw_pdbs/split_chain/1csn_A.pdb raw_pdbs/split_chain/1lp4_A.pdb ........ Extracting sequences pdb/seq: 1 name: raw_pdbs/split_chain/4b7t_A.pdb pdb/seq: 2 name: raw_pdbs/split_chain/2exm_A.pdb pdb/seq: 3 name: raw_pdbs/split_chain/1opj_A.pdb pdb/seq: 4 name: raw_pdbs/split_chain/4jaj_A.pdb pdb/seq: 5 name: raw_pdbs/split_chain/1a9u_A.pdb pdb/seq: 6 name: raw_pdbs/split_chain/1tki_A.pdb pdb/seq: 7 name: raw_pdbs/split_chain/1csn_A.pdb pdb/seq: 8 name: raw_pdbs/split_chain/1lp4_A.pdbsummary(c(seqidentity(pdbs)))Min. 1st Qu. Median Mean 3rd Qu. Max. 0.1520 0.2010 0.2530 0.3402 0.3150 1.0000# NMA on all structures modes <- nma.pdbs(pdbs, ncore=NULL)Details of Scheduled Calculation: ... 8 input structures ... storing 711 eigenvectors for each structure ... dimension of x$U.subspace: ( 717x711x8 ) ... coordinate superposition prior to NM calculation ... aligned eigenvectors (gap containing positions removed) ... estimated memory usage of final 'eNMA' object: 31.2 Mb | | | 0%# Calculate correlation matrices for each structure cij <- dccm(modes) # Set DCCM plot panel names for combined figure dimnames(cij$all.dccm) = list(NULL, NULL, ids) plot.dccm(cij$all.dccm) # Filter to display only correlations present in all structures cij.all <- filter.dccm(cij, cutoff.sims = 8, cutoff.cij = 0)
plot.dccm(cij.all, main = "Consensus Residue Cross Correlation")
detach(transducin)