DCCM Plot
Usage
"plot"(x, resno=NULL, sse=NULL, colorkey=TRUE, at=c(-1, -0.75, -0.5, -0.25, 0.25, 0.5, 0.75, 1), main="Residue Cross Correlation", helix.col = "gray20", sheet.col = "gray80", inner.box=TRUE, outer.box=FALSE, xlab="Residue No.", ylab="Residue No.", margin.segments=NULL, segment.col=vmd_colors(), segment.min=1, ...)
Arguments
- x
- a numeric matrix of atom-wise cross-correlations as output by the ‘dccm’ function.
- resno
- an optional vector with length equal to that of
xthat will be used to annotate the x- and y-axis. This is typically a vector of residue numbers. Can be also provided with a ‘pdb’ object, in which ‘resno’ of all C-alpha atoms will be used. If NULL residue positions from 1 to the length ofxwill be used. See examples below. - sse
- secondary structure object as returned from
dssp,strideorread.pdb. - colorkey
- logical, if TRUE a key is plotted.
- at
- numeric vector specifying the levels to be colored.
- main
- a main title for the plot.
- helix.col
- The colors for rectangles representing alpha helices.
- sheet.col
- The colors for rectangles representing beta strands.
- inner.box
- logical, if TRUE an outer box is drawn.
- outer.box
- logical, if TRUE an outer box is drawn.
- xlab
- a label for the x axis.
- ylab
- a label for the y axis.
- margin.segments
- a numeric vector of cluster membership as obtained from cutree() or other community detection method. This will be used for bottom and left margin annotation.
- segment.col
- a vector of colors used for each cluster group in margin.segments.
- segment.min
- a single element numeric vector that will cause margin.segments with a length below this value to be excluded from the plot.
- ...
- additional graphical parameters for contourplot.
Description
Plot a dynamical cross-correlation matrix.
Details
See the ‘contourplot’ function from the lattice package for plot customization options, and the functions dssp and stride for further details.
Value
-
Called for its effect.
References
Grant, B.J. et al. (2006) Bioinformatics 22, 2695--2696.
Note
Be sure to check the correspondence of your ‘sse’ object with the ‘cij’ values being plotted as no internal checks are currently performed.
Examples
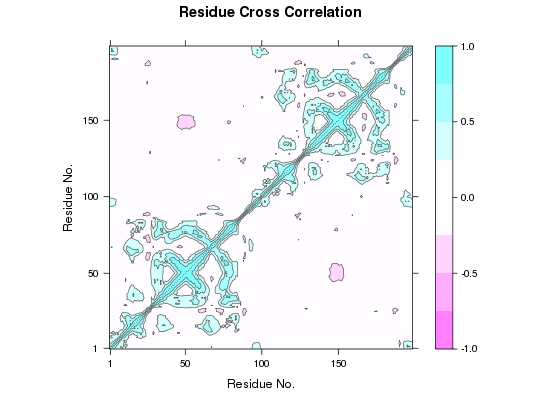
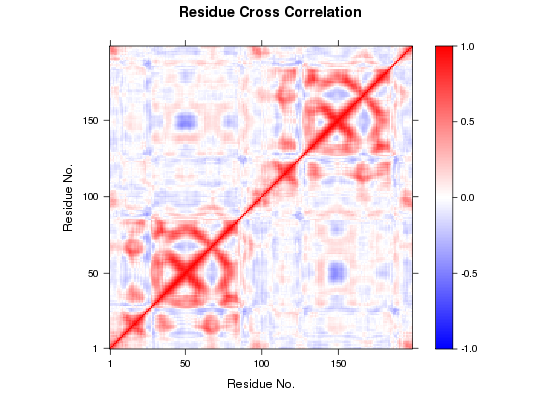
##-- Read example trajectory file trtfile <- system.file("examples/hivp.dcd", package="bio3d") trj <- read.dcd(trtfile)NATOM = 198 NFRAME= 117 ISTART= 0 last = 117 nstep = 117 nfile = 117 NSAVE = 1 NDEGF = 0 version 24 Reading (x100) |======================================================================| 100%## Read reference PDB and trim it to match the trajectory pdb <- trim(read.pdb("1W5Y"), 'calpha')Note: Accessing on-line PDB file## select residues 24 to 27 and 85 to 90 in both chains inds <- atom.select(pdb, resno=c(24:27,85:90)) ## lsq fit of trj on pdb xyz <- fit.xyz(pdb$xyz, trj, fixed.inds=inds$xyz, mobile.inds=inds$xyz) ## Dynamic cross-correlations of atomic displacements cij <- dccm(xyz) ## Default plot plot.dccm(cij) ## Change the color scheme and the range of colored data levels plot.dccm(cij, contour=FALSE, col.regions=bwr.colors(200), at=seq(-1,1,by=0.01) )
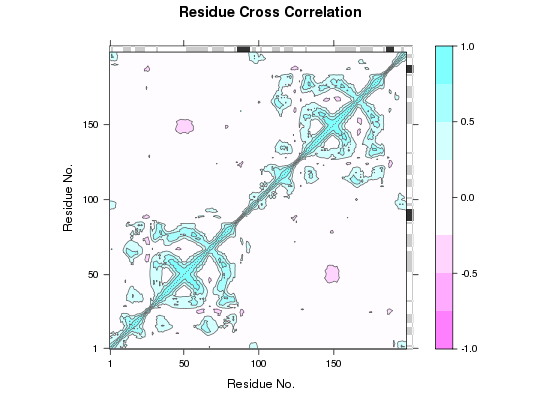
## Add secondary structure annotation to plot margins plot.dccm(cij, sse=pdb)
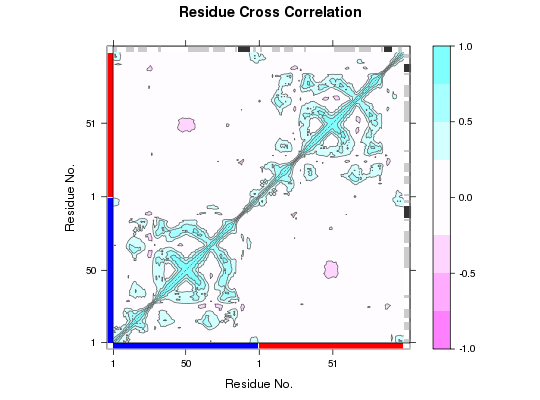
## Add additional margin annotation for chains ## Also label x- and y-axis with PDB residue numbers ch <- ifelse(pdb$atom$chain=="A", 1,2) plot.dccm(cij, resno=pdb, sse=pdb, margin.segments=ch)
## Plot with cluster annotation from dynamic network analysis #net <- cna(cij) #plot.dccm(cij, margin.segments=net$raw.communities$membership) ## Focus on major communities (i.e. exclude those below a certain total length) #plot.dccm(cij, margin.segments=net$raw.communities$membership, segment.min=25)
See also
plot.bio3d, plot.dmat,
filled.contour, contour,
image plot.default, dssp,
stride