Overlap analysis
Usage
overlap(modes, dv, nmodes=20)
Arguments
- modes
- an object of class
"pca"or"nma"as obtained from functionpca.xyzornma. Alternatively a 3NxM matrix of eigenvectors can be provided. - dv
- a displacement vector of length 3N.
- nmodes
- the number of modes in which the calculation should be based.
Description
Calculate the squared overlap between sets of vectors.
Details
Squared overlap (or dot product) is used to measure the similiarity between a displacement vector (e.g. a difference vector between two conformational states) and mode vectors obtained from principal component or normal modes analysis.
By definition the cumulative sum of the overlap values equals to one.
Structure modes$U (or alternatively, the 3NxM matrix of eigenvectors)
should be of same length (3N) as dv.
Value
-
Returns a list with the following components:
- overlap
- a numeric vector of the squared dot products (overlap values)
between the (normalized) vector (
dv) and each mode inmode. - overlap.cum
- a numeric vector of the cumulative squared overlap values.
References
Skjaerven, L. et al. (2011) Proteins 79, 232--243. Grant, B.J. et al. (2006) Bioinformatics 22, 2695--2696.
Examples
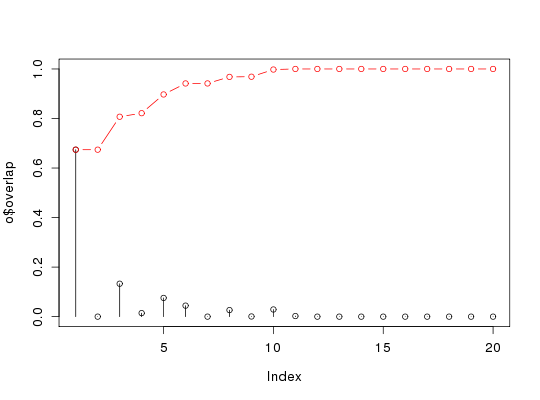
attach(kinesin) # Ignore gap containing positions ##gaps.res <- gap.inspect(pdbs$ali) gaps.pos <- gap.inspect(pdbs$xyz) #-- Do PCA pc.xray <- pca.xyz(pdbs$xyz[, gaps.pos$f.inds]) # Define a difference vector between two structural states diff.inds <- c(grep("d1v8ka", pdbs$id), grep("d1goja", pdbs$id)) dv <- difference.vector( pdbs$xyz[diff.inds,], gaps.pos$f.inds ) # Calculate the squared overlap between the PCs and the difference vector o <- overlap(pc.xray, dv) o <- overlap(pc.xray$U, dv) # Plot results plot(o$overlap, type='h', ylim=c(0,1)) points(o$overlap)lines(o$overlap.cum, type='b', col='red')
detach(kinesin) ## Calculate overlap from NMA pdb.a <- read.pdb("1cmk")Note: Accessing on-line PDB file PDB has ALT records, taking A only, rm.alt=TRUEpdb.b <- read.pdb("3dnd")Note: Accessing on-line PDB file PDB has ALT records, taking A only, rm.alt=TRUE## Fetch CA coordinates sele.a <- atom.select(pdb.a, chain='E', resno=c(15:350), elety='CA') sele.b <- atom.select(pdb.b, chain='A', resno=c(1:350), elety='CA') xyz <- rbind(pdb.a$xyz[sele.a$xyz], pdb.b$xyz[sele.b$xyz]) ## Superimpose xyz[2,] <- fit.xyz(xyz[1,], xyz[2,], 1:ncol(xyz)) ## The difference between the two conformations dv <- difference.vector( xyz ) ## Calculate normal modes modes <- nma(pdb.a, inds=sele.a)Building Hessian... Done in 0.187 seconds. Diagonalizing Hessian... Done in 2.028 seconds.# Calculate the squared overlap between the normal modes # and the difference vector o <- overlap(modes, dv)