Normal Mode Analysis
Usage
nma(...)
Arguments
- ...
- arguments passed to the methods
nma.pdb, ornma.pdbs. For functionnma.pdbthis will include an object of classpdbas obtained from functionread.pdb. For functionnma.pdbsan object of classpdbsas obtained from functionpdbalnorread.fasta.pdb.
Description
Perform normal mode analysis (NMA) on either a single or an ensemble of protein structures.
Details
Normal mode analysis (NMA) is a computational approach for studying and characterizing protein flexibility. Current functionality entails normal modes calculation on either a single protein structure or an ensemble of aligned protein structures.
This generic nma function calls the corresponding
methods for the actual calculation, which is determined by the class
of the input argument:
Function nma.pdb will be used when the input argument is
of class pdb. The function calculates the normal modes of a
C-alpha model of a protein structure.
Function nma.pdbs will be used when the input argument is
of class pdbs. The function will perform normal mode analysis
of each PDB structure stored in the pdbs object
(‘ensemble NMA’).
See documentation and examples for each corresponding function for more details.
References
Skjaerven, L. et al. (2014) BMC Bioinformatics 15, 399. Grant, B.J. et al. (2006) Bioinformatics 22, 2695--2696.
Examples
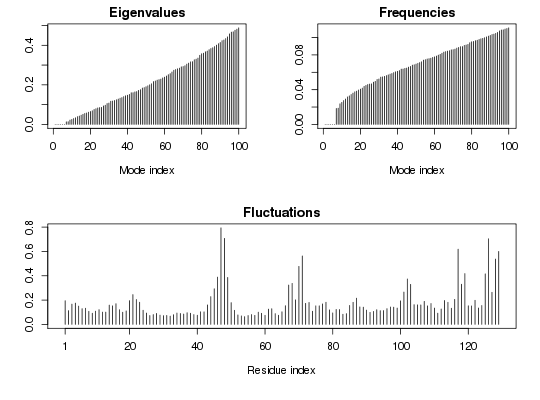
##- Singe structure NMA ## Fetch stucture pdb <- read.pdb( system.file("examples/1hel.pdb", package="bio3d") ) ## Calculate normal modes modes <- nma(pdb)Building Hessian... Done in 0.053 seconds. Diagonalizing Hessian... Done in 0.133 seconds.## Print modes print(modes)Call: nma.pdb(pdb = pdb) Class: VibrationalModes (nma) Number of modes: 387 (6 trivial) Frequencies: Mode 7: 0.018 Mode 8: 0.019 Mode 9: 0.024 Mode 10: 0.025 Mode 11: 0.028 Mode 12: 0.029 + attr: modes, frequencies, force.constants, fluctuations, U, L, xyz, mass, temp, triv.modes, natoms, call## Plot modes plot(modes) ## Visualize modes #m7 <- mktrj.nma(modes, mode=7, file="mode_7.pdb") ## Needs MUSCLE installed - testing excluded ##- Ensemble NMA ## Fetch PDB files and split to chain A only PDB files ids <- c("1a70_A", "1czp_A", "1frd_A", "1fxi_A", "1iue_A", "1pfd_A")
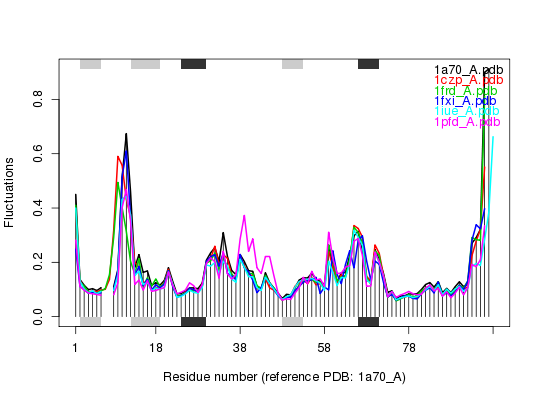
files <- get.pdb(ids, split = TRUE, path = tempdir())|======================================================================| 100%## Sequence Alignement pdbs <- pdbaln(files, outfile = tempfile())Reading PDB files: /tmp/RtmpTDihxb/split_chain/1a70_A.pdb /tmp/RtmpTDihxb/split_chain/1czp_A.pdb /tmp/RtmpTDihxb/split_chain/1frd_A.pdb /tmp/RtmpTDihxb/split_chain/1fxi_A.pdb /tmp/RtmpTDihxb/split_chain/1iue_A.pdb /tmp/RtmpTDihxb/split_chain/1pfd_A.pdb . PDB has ALT records, taking A only, rm.alt=TRUE ..... Extracting sequences pdb/seq: 1 name: /tmp/RtmpTDihxb/split_chain/1a70_A.pdb pdb/seq: 2 name: /tmp/RtmpTDihxb/split_chain/1czp_A.pdb PDB has ALT records, taking A only, rm.alt=TRUE pdb/seq: 3 name: /tmp/RtmpTDihxb/split_chain/1frd_A.pdb pdb/seq: 4 name: /tmp/RtmpTDihxb/split_chain/1fxi_A.pdb pdb/seq: 5 name: /tmp/RtmpTDihxb/split_chain/1iue_A.pdb pdb/seq: 6 name: /tmp/RtmpTDihxb/split_chain/1pfd_A.pdb## Normal mode analysis on aligned data modes <- nma(pdbs, rm.gaps=FALSE)Details of Scheduled Calculation: ... 6 input structures ... storing 282 eigenvectors for each structure ... dimension of x$U.subspace: ( 300x282x6 ) ... coordinate superposition prior to NM calculation ... estimated memory usage of final 'eNMA' object: 3.9 Mb |======================================================================| 100%## Plot fluctuation data plot(modes, pdbs=pdbs)Extracting SSE from pdbs$sse attribute