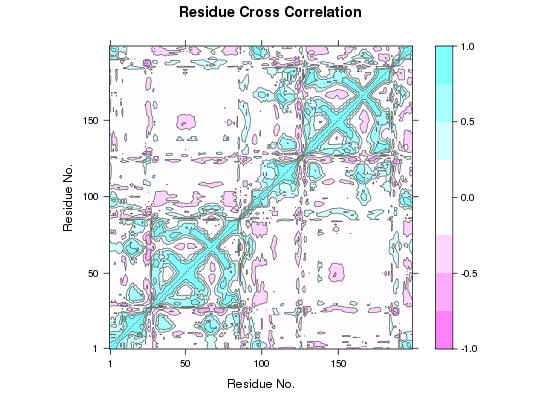
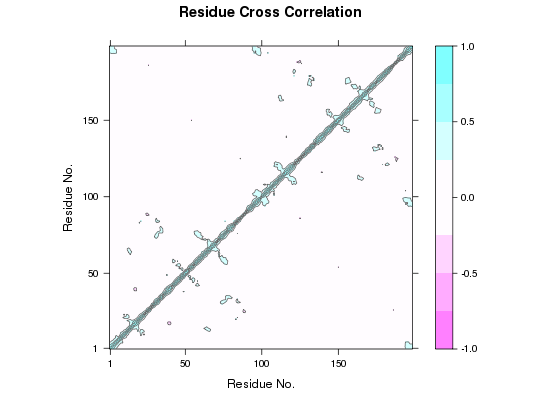
Dynamic Cross-Correlation from Principal Component Analysis
Usage
"dccm"(x, pc = NULL, ncore = NULL, ...)
Arguments
- x
- an object of class
pcaas obtained from functionpca.xyz. - pc
- numerical, indices of PCs to be included in the calculation.
If all negative, PCs complementary to
abs(pc)are included. - ncore
- number of CPU cores used to do the calculation.
By default (
ncore = NULL), use all available cores detected. - ...
- additional arguments to
cov2dccm.
Description
Calculate the cross-correlation matrix from principal component analysis (PCA).
Details
This function calculates the cross-correlation matrix from principal
component analysis (PCA) obtained from pca.xyz of a set of protein
structures. It is an alternative way to calculate correlation in addition
to the conventional way from xyz coordinates directly. But, in this new
way one can freely chooses the PCs to be included in the
calculation (e.g. filter PCs with small eigenvalues).
Value
-
Returns a cross-correlation matrix.
References
Grant, B.J. et al. (2006) Bioinformatics 22, 2695--2696.
Examples
##-- Read example trajectory file trtfile <- system.file("examples/hivp.dcd", package="bio3d") trj <- read.dcd(trtfile)NATOM = 198 NFRAME= 117 ISTART= 0 last = 117 nstep = 117 nfile = 117 NSAVE = 1 NDEGF = 0 version 24 Reading (x100) |======================================================================| 100%## Read the starting PDB file to determine atom correspondence pdbfile <- system.file("examples/hivp.pdb", package="bio3d") pdb <- read.pdb(pdbfile) ## Select residues 24 to 27 and 85 to 90 in both chains inds <- atom.select(pdb, resno=c(24:27,85:90), elety='CA') ## lsq fit of trj on pdb xyz <- fit.xyz(pdb$xyz, trj, fixed.inds=inds$xyz, mobile.inds=inds$xyz) ## Do PCA pca <- pca.xyz(xyz) ## DCCM: only use first 10 PCs cij <- dccm(pca, pc = c(1:10)) ## Plot DCCM plot(cij) ## DCCM: remove first 10 PCs cij <- dccm(pca, pc = -c(1:10))
## Plot DCCM plot(cij)