Gaussian Network Model
Usage
gnm(x, ...)"gnm"(x, inds = NULL, temp = 300, keep = NULL, outmodes = NULL, gamma = 1, cutoff = 8, check.connect = TRUE, ...)"gnm"(x, fit = TRUE, full = FALSE, subspace = NULL, rm.gaps = TRUE, gc.first = TRUE, ncore = NULL, ...)
Arguments
- x
- an object of class
pdbas obtained from functionread.pdb. - ...
- (in
gnm.pdbs) additional arguments passed tognm.pdb. - inds
- atom and xyz coordinate indices obtained from
atom.selectthat selects the elements ofpdbupon which the calculation should be based. If not provided the function will attempt to select all calpha atoms automatically. - temp
- numerical, temperature for which the amplitudes for scaling the atomic displacement vectors are calculated. Set ‘temp=NULL’ to avoid scaling.
- keep
- numerical, final number of modes to be stored. Note that all subsequent analyses are limited to this subset of modes. This option is useful for very large structures and cases where memory may be limited.
- outmodes
- atom indices as obtained from
atom.selectspecifying the atoms to include in the resulting mode object. - gamma
- numerical, global scale of the force constant.
- cutoff
- numerical, distance cutoff for pair-wise interactions.
- check.connect
- logical, if TRUE check chain connectivity.
- fit
- logical, if TRUE C-alpha coordinate based superposition is performed prior to normal mode calculations.
- full
- logical, if TRUE return the complete, full structure, ‘nma’ objects.
- subspace
- number of eigenvectors to store for further analysis.
- rm.gaps
- logical, if TRUE obtain the hessian matrices for only atoms in the aligned positions (non-gap positions in all aligned structures). Thus, gap positions are removed from output.
- gc.first
- logical, if TRUE will call gc() first before mode calculation
for each structure. This is to avoid memory overload when
ncore > 1. - ncore
- number of CPU cores used to do the calculation.
Value
-
Returns an object of class ‘gnm’ with the following components:
- force.constants
- numeric vector containing the force constants corresponding to each mode.
- fluctuations
- numeric vector of atomic fluctuations.
- U
- numeric matrix with columns containing the raw eigenvectors.
- L
- numeric vector containing the raw eigenvalues.
- xyz
- numeric matrix of class
xyzcontaining the Cartesian coordinates in which the calculation was performed. - temp
- numerical, temperature for which the amplitudes for scaling the atomic displacement vectors are calculated.
- triv.modes
- number of trivial modes.
- natoms
- number of C-alpha atoms.
- call
- the matched call.
Description
Perform Gaussian network model (GNM) based normal mode analysis (NMA) for a protein structure.
Details
This function builds a Gaussian network model (an isotropic elastic network model) for C-alpha atoms and performs subsequent normal mode analysis (NMA). The model employs a distance cutoff for the network construction: Atom pairs with distance falling within the cutoff have a harmonic interaction with a uniform force constant; Otherwise atoms have no interaction. Output contains N-1 (N, the number of residues) non-trivial modes (i.e. the degree of freedom is N-1), which can then be used to calculate atomic fluctuations and covariance.
References
Bahar, I. et al. (1997) Folding Des. 2, 173.
Examples
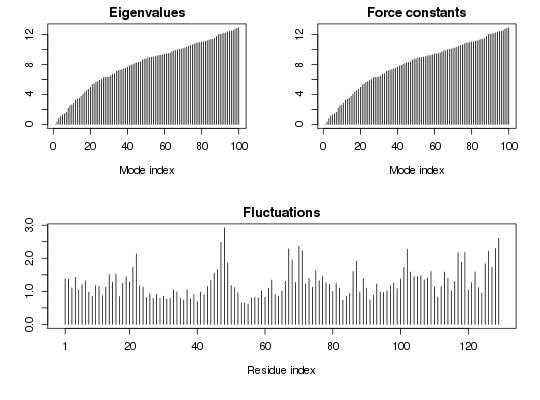
## Fetch stucture pdb <- read.pdb( system.file("examples/1hel.pdb", package="bio3d") ) ## Calculate normal modes modes <- gnm(pdb) ## Print modes print(modes)Call: gnm.pdb(x = pdb) Class: EnergeticModes (gnm) Number of modes: 129 (1 trivial) Force constants: Mode 2: 0.342 Mode 3: 0.804 Mode 4: 1.108 Mode 5: 1.277 Mode 6: 1.416 Mode 7: 1.617 + attr: force.constants, fluctuations, U, L, xyz, temp, triv.modes, natoms, call## Plot modes plot(modes)