
Methods for Computer Aided Drug Design
29/09/14 14:53
Current drug development strategies take one or a few rigid structure(s) of an individual protein and design a single molecule to block its activity (i.e., catalytic pocket). We are developing a more sophisticated methodology that exploits flexible target structures, allosteric pocket identification and evolutionary analysis.
Although ligand flexibility is now routinely accounted for in modern computational drug design, incorporating receptor flexibility remains an important challenge. I have recently adapted efficient conformational sampling methods to generate reliable receptor ensemble structures for use in drug discovery protocols.
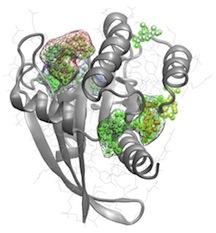
These enhanced sampling methods have also proven successful at identifying distal portions of protein structures that undergo correlated motions indicative of allosteric coupling. I plan to merge these methods with the extensive mutational data emerging from next-generation sequencing efforts. This work will utilize and build upon my successful Bio3D software infrastructure. Bio3D has the unique capability of analyzing structural variability, correlated motions and large amounts of sequence data in a single integrated environment. The current version of Bio3D has been downloaded by over 13,700 researchers and cited 23 times in the last two years. Merging these new methods represents an important advance that will greatly facilitate the design of selective inhibitors.
<< Back to Research Overview
Although ligand flexibility is now routinely accounted for in modern computational drug design, incorporating receptor flexibility remains an important challenge. I have recently adapted efficient conformational sampling methods to generate reliable receptor ensemble structures for use in drug discovery protocols.
These enhanced sampling methods have also proven successful at identifying distal portions of protein structures that undergo correlated motions indicative of allosteric coupling. I plan to merge these methods with the extensive mutational data emerging from next-generation sequencing efforts. This work will utilize and build upon my successful Bio3D software infrastructure. Bio3D has the unique capability of analyzing structural variability, correlated motions and large amounts of sequence data in a single integrated environment. The current version of Bio3D has been downloaded by over 13,700 researchers and cited 23 times in the last two years. Merging these new methods represents an important advance that will greatly facilitate the design of selective inhibitors.
<< Back to Research Overview