Calculate Mainchain and Sidechain Torsion/Dihedral Angles
Usage
torsion.pdb(pdb)
Arguments
- pdb
- a PDB structure object as obtained from
function
read.pdb.
Description
Calculate all torsion angles for a given protein PDB structure object.
Details
The conformation of a polypeptide chain can be usefully described in
terms of angles of internal rotation around its constituent bonds. See
the related torsion.xyz function, which is called by this
function, for details.
Value
-
Returns a list object with the following components:
- phi
- main chain torsion angle for atoms C,N,CA,C.
- psi
- main chain torsion angle for atoms N,CA,C,N.
- omega
- main chain torsion angle for atoms CA,C,N,CA.
- alpha
- virtual torsion angle between consecutive C-alpha atoms.
- chi1
- side chain torsion angle for atoms N,CA,CB,*G.
- chi2
- side chain torsion angle for atoms CA,CB,*G,*D.
- chi3
- side chain torsion angle for atoms CB,*G,*D,*E.
- chi4
- side chain torsion angle for atoms *G,*D,*E,*Z.
- chi5
- side chain torsion angle for atoms *D,*E,*Z, NH1.
- coords
- numeric matrix of ‘justified’ coordinates.
- tbl
- a numeric matrix of psi, phi and chi torsion angles.
References
Grant, B.J. et al. (2006) Bioinformatics 22, 2695--2696.
Note
For the protein backbone, or main-chain atoms, the partial double-bond character of the peptide bond between ‘C=N’ atoms severely restricts internal rotations. In contrast, internal rotations around the single bonds between ‘N-CA’ and ‘CA-C’ are only restricted by potential steric collisions. Thus, to a good approximation, the backbone conformation of each residue in a given polypeptide chain can be characterised by the two angles phi and psi.
Sidechain conformations can also be described by angles of internal rotation denoted chi1 up to chi5 moving out along the sidechain.
Examples
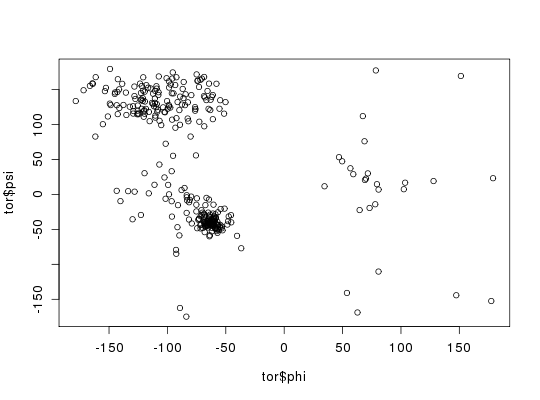
# PDB server connection required - testing excluded ##-- PDB torsion analysis pdb <- read.pdb( "1bg2" )Note: Accessing on-line PDB filetor <- torsion.pdb(pdb) head(tor$tbl)phi psi chi1 chi2 chi3 chi4 chi5 [1,] NA -21.550499 -45.13716 -47.35099 NA NA NA [2,] -64.02592 -59.916666 175.35429 58.11471 NA NA NA [3,] -140.22879 -9.607914 NA NA NA NA NA [4,] 67.34903 112.010164 -141.68005 -60.79993 -52.19896 NA NA [5,] 68.79209 76.000250 179.25704 NA NA NA NA [6,] -95.17049 55.249681 -54.67200 29.83050 NA NA NA## basic Ramachandran plot plot(tor$phi, tor$psi) ## torsion analysis of a single coordinate vector #inds <- atom.select(pdb,"calpha") #tor.ca <- torsion.xyz(pdb$xyz[inds$xyz], atm.inc=1) ##-- Compare two PDBs to highlight interesting residues aln <- read.fasta(system.file("examples/kif1a.fa",package="bio3d"))
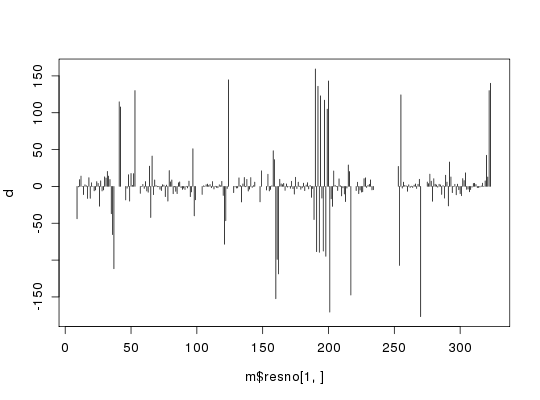
m <- read.fasta.pdb(aln)pdb/seq: 1 name: http://www.rcsb.org/pdb/files/1bg2.pdb pdb/seq: 2 name: http://www.rcsb.org/pdb/files/1i6i.pdb PDB has ALT records, taking A only, rm.alt=TRUE pdb/seq: 3 name: http://www.rcsb.org/pdb/files/1i5s.pdb PDB has ALT records, taking A only, rm.alt=TRUE pdb/seq: 4 name: http://www.rcsb.org/pdb/files/2ncd.pdba <- torsion.xyz(m$xyz[1,],1) b <- torsion.xyz(m$xyz[2,],1) d <- wrap.tor(a-b) plot(m$resno[1,],d, typ="h")