Atomic RMS Fluctuations
Usage
rmsf(xyz, grpby=NULL, average=FALSE)
Arguments
- xyz
- numeric matrix of coordinates with each row corresponding to an individual conformer.
- grpby
- a vector counting connective duplicated elements that indicate the elements of 'xyz' that should be considered as a group (e.g. atoms from a particular residue). If provided a 'pdb' object, grouping is automatically set by amino acid residues.
- average
- logical, if TRUE averaged over atoms.
Description
Calculate atomic root mean squared fluctuations.
Details
RMSF is an often used measure of conformational variance. It is calculated by
f_i=sqrt((1/(M-1))Sum_j(||r_i^j-r_i^0||^2)),
where f_i is the RMSF value for the ith atom, M the total number of frames
(total number of rows of xyz), r_i^j the positional vector of the
ith atom in the jth frame, and r_i^0 the mean position of ith atom.
||r|| denotes the Euclidean norm of the vector r.
Value
-
Returns a numeric vector of RMSF values. If
average=TRUE a single numeric value
representing the averaged RMSF value over all atoms will be returned.
References
Grant, B.J. et al. (2006) Bioinformatics 22, 2695--2696.
Examples
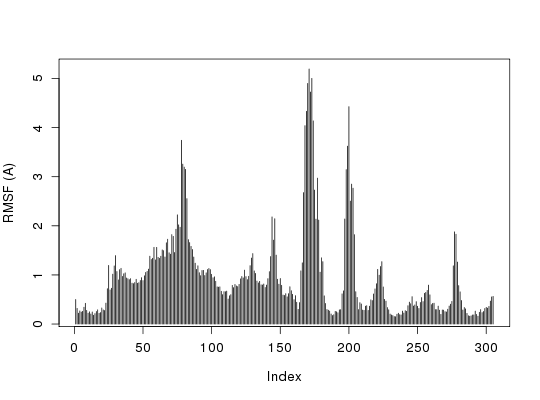
attach(transducin)The following objects are masked from transducin (pos = 3): annotation, core, pdbs# Ignore Gaps gaps <- gap.inspect(pdbs$ali) r <- rmsf(pdbs$xyz) plot(r[gaps$f.inds], typ="h", ylab="RMSF (A)") detach(transducin)
pdb <- read.pdb("1d1d", multi=TRUE)Note: Accessing on-line PDB filexyz <- pdb$xyz # superimpose trajectory xyz <- fit.xyz(xyz[1, ], xyz)Warning message: No fitting indices provided, using the 3395 non NA positions# select mainchain atoms sele <- atom.select(pdb, elety=c("CA", "C", "N", "O")) # residue numbers to group by resno <- pdb$atom$resno[sele$atom] # mean rmsf value of mainchain atoms of each residue r <- rmsf(xyz[, sele$xyz], grpby=resno) plot.bio3d(r, resno=pdb, sse=pdb, ylab="RMSF (A)")