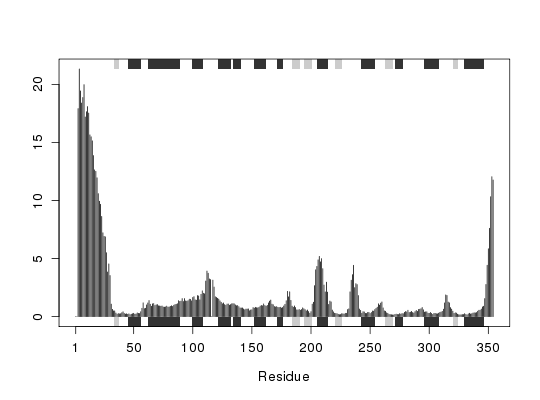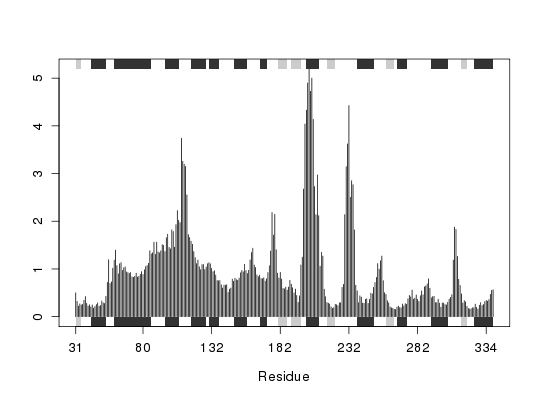SSE annotation for a PDBs Object
Usage
pdbs2sse(pdbs, ind = NULL, rm.gaps = TRUE, resno = TRUE, pdb = FALSE, ...)
Arguments
- pdbs
- a list of class
"pdbs"containing PDB file data, as obtained fromread.fasta.pdborpdbaln. - ind
- numeric index pointing to the PDB in which the SSE should
be provided. If
ind=NULL, then the consensus SSE is returned. - rm.gaps
- logical, if TRUE SSEs spanning gap containing columns are
omitted from the output in the resulting
sseobject. - resno
- logical, if TRUE output is in terms of residue numbers rather than residue index (position in sequence).
- pdb
- logical, if TRUE function
dsspwill be called on the correspondingpdbobject rather than to usepdbs$sseto obtain the SSE object. - ...
- arguments passed to function
dssp.
Description
Returns secondary structure element (SSE) annotation ("sse" object) for a structure in the provided "pdbs" object.
Details
This function provides a "sse" list object containing
secondary structure elements (SSE) annotation data for a particular
structure in the provided "pdbs" object. Residue numbers are
provided relative to the alignment in the "pdbs" object.
When ind=NULL the function will attemt to return the consensus
SSE annotation, i.e. where there are SSEs across all structures. This
will only work SSE data is found in the "pdbs" object.
See examples for more details.
Value
-
Returns a list object of class
sse.
References
Grant, B.J. et al. (2006) Bioinformatics 22, 2695--2696.
Examples
attach(transducin) ## calculate RMSF rf <- rmsf(pdbs$xyz) ## Fetch SSE annotation, output in terms of alignment index sse <- pdbs2sse(pdbs, ind=1, rm.gaps=FALSE, resno=FALSE)Extracting SSE from pdbs$sse attribute## Add SSE annotation to plot plotb3(rf, sse=sse) ## Calculate RMSF only for non-gap columns gaps.pos <- gap.inspect(pdbs$xyz)
rf <- rmsf(pdbs$xyz[, gaps.pos$f.inds]) ## With gap columns removed, output in terms of residue number sse <- pdbs2sse(pdbs, ind=1, rm.gaps=TRUE, resno=TRUE)Extracting SSE from pdbs$sse attributegaps.res <- gap.inspect(pdbs$ali) plotb3(rf, sse=sse, resno=pdbs$resno[1, gaps.res$f.inds])
detach(transducin)