Mask a Subset of Atoms in a DCCM Object.
Usage
mask(...)"mask"(dccm, pdb = NULL, a.inds = NULL, b.inds = NULL, ...)
Arguments
- dccm
- a DCCM structure object obtained from function
dccm. - pdb
- a PDB structure object obtained from
read.pdb. Must match the dimensions ofdccm. - a.inds
- a numeric vector containing the indices of the elements
of the DCCM matrix in which should not be masked. Alternatively, if
pdbis provided a selection object (as obtained fromatom.select) can be provided. - b.inds
- a numeric vector containing the indices of the elements of the DCCM matrix in which should not be masked.
- ...
- arguments not passed anywhere.
Description
Produce a new DCCM object with selected atoms masked.
Details
This is a basic utility function for masking a DCCM object matrix to highlight user-selected regions in the correlation network.
When both a.inds and b.inds are provided only their
intersection is retained. When only a.inds is provided then
the corresponding region to everything else is retained.
Note: The current version assumes that the input PDB corresponds to the input DCCM. In many cases this will correspond to a PDB object containing only CA atoms.
Value
-
Returns a matrix list of class
"dccm" with the indices/atoms
not corresponding to the selection masked.
References
Grant, B.J. et al. (2006) Bioinformatics 22, 2695--2696.
Examples
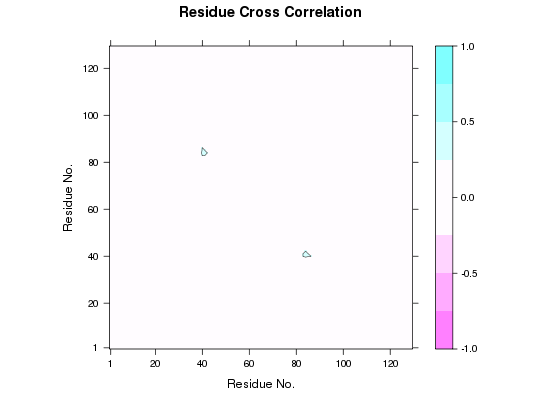
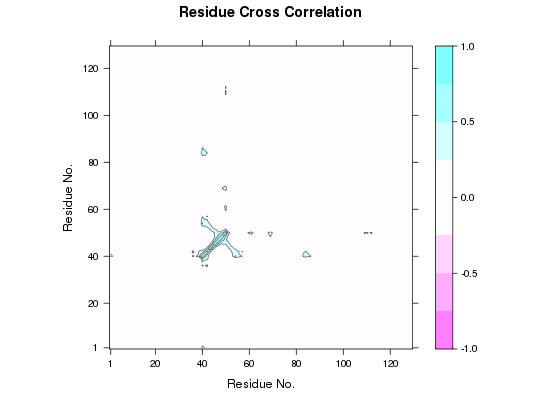
## Calculate DCCM pdb <- read.pdb( system.file("examples/1hel.pdb", package="bio3d") ) cij <- dccm(nma(pdb))Building Hessian... Done in 0.047 seconds. Diagonalizing Hessian... Done in 0.132 seconds. |======================================================================| 100%## Mask DCCM matrix according to matrix indices cijm <- mask(cij, a.inds=40:50, b.inds=80:90) plot(cijm) ## Retain only 40:50 to everything else cijm <- mask(cij, a.inds=40:50)
plot(cijm)
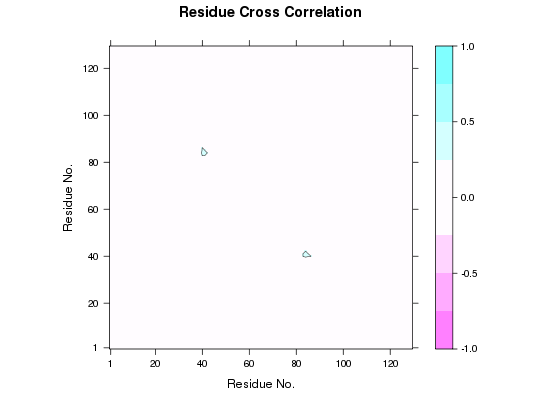
## Mask DCCM matrix according PDB selection pdb.ca <- trim(pdb, "calpha") a.inds <- atom.select(pdb.ca, resno=40:50) b.inds <- atom.select(pdb.ca, resno=80:90) # Provide pdb object correspoding to input dccm cijm <- mask(cij, pdb.ca, a.inds, b.inds) plot(cijm)