RMSD Filter
Usage
filter.rmsd(xyz = NULL, rmsd.mat = NULL, cutoff = 0.5, fit = TRUE, verbose = TRUE, inds = NULL, method = "complete", ...)
Arguments
- xyz
- a numeric matrix or list object containing multiple
coordinates for pairwise comparison, such as that obtained from
read.fasta.pdb. Not used ifrmsd.matis given. - rmsd.mat
- an optional matrix of RMSD values obtained from
rmsd. - cutoff
- a numeric rmsd cutoff value.
- fit
- logical, if TRUE coordinate superposition is performed prior to RMSD calculation.
- verbose
- logical, if TRUE progress details are printed.
- inds
- a vector of indices that selects the elements of
xyzupon which the calculation should be based. By default, all the non-gap sites inxyz. - method
- the agglomeration method to be used. See function
hclustfor more information. - ...
- additional arguments passed to and from functions.
Description
Identify and filter subsets of conformations at a given RMSD cutoff.
Details
This function performs hierarchical cluster analysis of a given matrix of RMSD values ‘rmsd.mat’, or an RMSD matrix calculated from a given coordinate matrix ‘xyz’, to identify conformers that fall below a given RMSD cutoff value ‘cutoff’.
Value
-
Returns a list object with components:
- ind
- indices of the conformers (rows) below the cutoff value.
- tree
- an object of class
"hclust", which describes the tree produced by the clustering process. - rmsd.mat
- a numeric matrix with all pairwise RMSD values.
References
Grant, B.J. et al. (2006) Bioinformatics 22, 2695--2696.
Examples
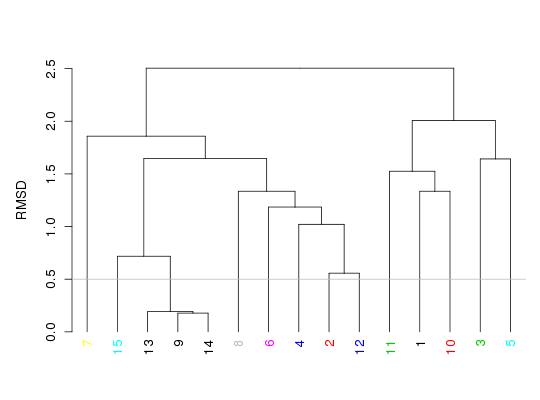
attach(kinesin) k <- filter.rmsd(xyz=pdbs,cutoff=0.5)filter.rmsd(): N clusters @ cutoff = 13pdbs$id[k$ind][1] "d1bg2__" "d2kin.1" "d1v8ka_" "d1goja_" "d1ry6a_" "d1sdma_" "d1t5c_a" [8] "d1vfva_" "d2fky_a" "d2ncda_" "d1f9va_" "d1mkja_" "d2fme_a"hclustplot(k$tree, h=0.5, ylab="RMSD") abline(h=0.5, col="gray")
detach(kinesin)