Distance Matrix Analysis
Usage
dm(...)"dm"(pdb, inds = NULL, grp = TRUE, verbose=TRUE, ...) "dm"(pdbs, ...)"dm"(xyz, grpby = NULL, scut = NULL, mask.lower = TRUE, gc.first=FALSE, ncore=1, ...)
Arguments
- pdb
- a
pdbstructure object as returned byread.pdbor a numeric vector of ‘xyz’ coordinates. - inds
- atom and xyz coordinate indices obtained from
atom.selectthat selects the elements ofpdbupon which the calculation should be based. - grp
- logical, if TRUE atomic distances will be grouped according to their residue membership. See ‘grpby’.
- verbose
- logical, if TRUE possible warnings are printed.
- pdbs
- a ‘pdbs’ object as returned by
read.fasta.pdborpdbaln. - xyz
- a numeric vector or matrix of Cartesian coordinates.
- grpby
- a vector counting connective duplicated elements that
indicate the elements of
xyzthat should be considered as a group (e.g. atoms from a particular residue). - scut
- a cutoff neighbour value which has the effect of excluding atoms, or groups, that are sequentially within this value.
- mask.lower
- logical, if TRUE the lower matrix elements (i.e. those below the diagonal) are returned as NA.
- gc.first
- logical, if TRUE will call gc() first before calculation of
distance matrix. This is to solve the memory overload problem when
ncore > 1andxyzhas many rows/columns, with a bit sacrifice on speed. - ncore
- number of CPU cores used to do the calculation.
ncore>1requires package ‘parallel’ installed. - ...
- arguments passed to and from functions.
Description
Construct a distance matrix for a given protein structure.
Details
Distance matrices, also called distance plots or distance maps, are an established means of describing and comparing protein conformations (e.g. Phillips, 1970; Holm, 1993).
A distance matrix is a 2D representation of 3D structure that is independent of the coordinate reference frame and, ignoring chirality, contains enough information to reconstruct the 3D Cartesian coordinates (e.g. Havel, 1983).
Value
-
Returns a numeric matrix of class
"dmat", with all N by N
distances, where N is the number of selected atoms. With multiple
frames the output is provided in a three dimensional array.
References
Grant, B.J. et al. (2006) Bioinformatics 22, 2695--2696.
Phillips (1970) Biochem. Soc. Symp. 31, 11--28.
Holm (1993) J. Mol. Biol. 233, 123--138.
Havel (1983) Bull. Math. Biol. 45, 665--720.
Note
The input selection can be any character string or pattern
interpretable by the function atom.select. For example,
shortcuts "calpha", "back", "all" and selection
strings of the form /segment/chain/residue number/residue
name/element number/element name/; see atom.select
for details.
If a coordinate vector is provided as input (rather than a pdb
object) the selection option is redundant and the input vector
should be pruned instead to include only desired positions.
Examples
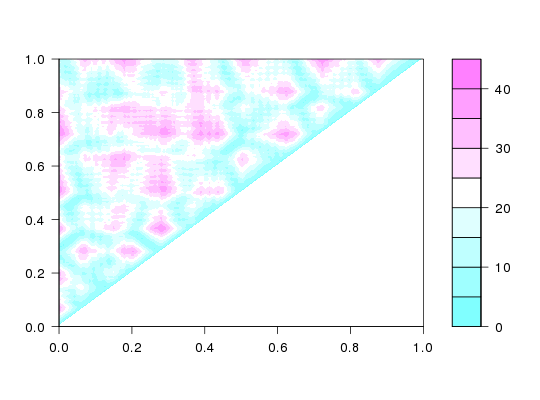
# PDB server connection required - testing excluded ##--- Distance Matrix Plot pdb <- read.pdb( "4q21" )Note: Accessing on-line PDB filek <- dm(pdb,inds="calpha") filled.contour(k, nlevels = 10) ## NOTE: FOLLOWING EXAMPLE NEEDS MUSCLE INSTALLED ##--- DDM: Difference Distance Matrix # Downlaod and align two PDB files pdbs <- pdbaln( get.pdb( c( "4q21", "521p"), path = tempdir() ), outfile = tempfile() )Warning message: /tmp/RtmpTDihxb/4q21.pdb exists. Skipping download
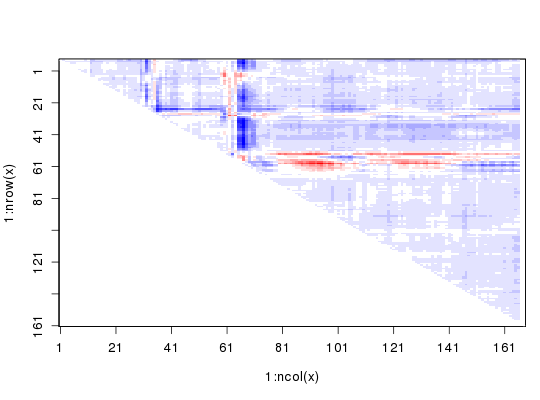
Reading PDB files: /tmp/RtmpTDihxb/4q21.pdb /tmp/RtmpTDihxb/521p.pdb .. Extracting sequences pdb/seq: 1 name: /tmp/RtmpTDihxb/4q21.pdb pdb/seq: 2 name: /tmp/RtmpTDihxb/521p.pdb# Get distance matrix a <- dm.xyz(pdbs$xyz[1,]) b <- dm.xyz(pdbs$xyz[2,]) # Calculate DDM c <- a - b # Plot DDM plot(c,key=FALSE, grid=FALSE)
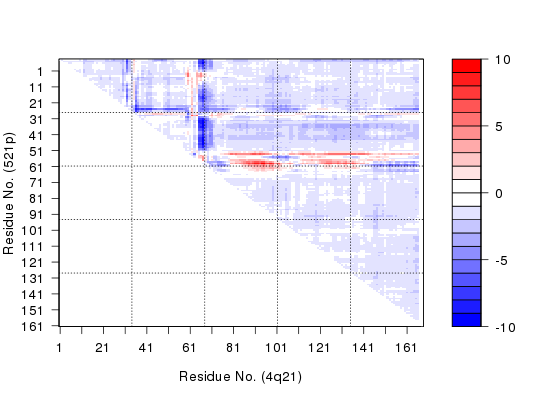
plot(c, axis.tick.space=10, resnum.1=pdbs$resno[1,], resnum.2=pdbs$resno[2,], grid.col="black", xlab="Residue No. (4q21)", ylab="Residue No. (521p)")
##-- Residue-wise distance matrix based on the ## minimal distance between all available atoms l <- dm.xyz(pdb$xyz, grpby=pdb$atom[,"resno"], scut=3)